the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 16 Jun 2020
| 16 Jun 2020
Variability and past long-term changes of brominated very short-lived substances at the tropical tropopause
Susann Tegtmeier
Elliot Atlas
Birgit Quack
Franziska Ziska
Kirstin Krüger
Halogenated very short-lived substances (VSLSs), such as bromoform (CHBr3), can be transported to the stratosphere and contribute to the halogen loading and ozone depletion. Given their highly variable emission rates and their short atmospheric lifetimes, the exact amount as well as the spatio-temporal variability of their contribution to the stratospheric halogen loading are still uncertain. We combine observational data sets with Lagrangian atmospheric modelling in order to analyse the spatial and temporal variability of the CHBr3 injection into the stratosphere for the time period 1979–2013. Regional maxima with mixing ratios of up to 0.4–0.5 ppt at 17 km altitude are diagnosed to be over Central America (1) and over the Maritime Continent–west Pacific (2), both of which are confirmed by high-altitude aircraft campaigns. The CHBr3 maximum over Central America is caused by the co-occurrence of convectively driven short transport timescales and strong regional sources, which in conjunction drive the seasonality of CHBr3 injection. Model results at a daily resolution reveal isolated, exceptionally high CHBr3 values in this region which are confirmed by aircraft measurements during the ACCENT campaign and do not occur in spatially or temporally averaged model fields. CHBr3 injection over the west Pacific is centred south of the Equator due to strong oceanic sources underneath prescribed by the here-applied bottom-up emission inventory. The globally largest CHBr3 mixing ratios at the cold point level of up to 0.6 ppt are diagnosed to occur over the region of India, Bay of Bengal, and Arabian Sea (3); however, no data from aircraft campaigns are available to confirm this finding. Inter-annual variability of stratospheric CHBr3 injection of 10 %–20 % is to a large part driven by the variability of coupled ocean–atmosphere circulation systems. Long-term changes, on the other hand, correlate with the regional sea surface temperature trends resulting in positive trends of stratospheric CHBr3 injection over the west Pacific and Asian monsoon region and negative trends over the east Pacific. For the tropical mean, these opposite regional trends balance each other out, resulting in a relatively weak positive trend of 0.017±0.012 ppt Br per decade for 1979–2013, corresponding to 3 % Br per decade. The overall contribution of CHBr3 together with CH2Br2 to the stratospheric halogen loading accounts for 4.7 ppt Br, in good agreement with existing studies, with 50 % and 50 % being injected in the form of source and product gases, respectively.
- Article
(16320 KB) -
Supplement
(6921 KB) - BibTeX
- EndNote





It has long been recognized that the depletion of stratospheric ozone over the last 30 years is mainly caused by human-made chlorine- and bromine-containing substances, often referred to as ozone-depleting substances (ODSs) (Carpenter et al., 2014). The Montreal Protocol, crafted in 1987 to control the production and consumption of ODSs, has been very successful in reducing the emission of the long-lived halocarbons. As a result, the overall abundance of ODSs in the atmosphere has been decreasing since the beginning of the 21st century, and the stratospheric ozone layer is expected to recover around the middle of the 21st century (Austin and Butchart, 2003; Carpenter et al., 2014; Salawitch et al., 2019).
In contrast to long-lived halocarbons, the so-called very short-lived substances (VSLSs) with chemical lifetimes of less than 6 months (e.g. Ko et al., 2003) are not controlled by the Montreal Protocol and are even suggested to increase in the future (e.g. Pyle et al., 2007; Tegtmeier et al., 2015; Ziska et al., 2017). Brominated VSLSs are primarily of natural origin, emitted by oceanic macroalgae and phytoplankton (e.g. Quack and Wallace, 2003). Over the last years there has been increasing evidence from observational (e.g. Dorf et al., 2008; Sioris et al., 2006; McLinden et al., 2010; Brinckmann et al., 2012) and modelling (e.g. Warwick et al., 2006; Liang et al., 2010; Hossaini et al., 2012b, 2016; Tegtmeier et al., 2012) studies that VSLSs provide a significant contribution to stratospheric total bromine (Bry). Current estimates of this contribution are about 5 (3–7) ppt bromine (Engel et al., 2018; Navarro et al., 2015; Wales et al., 2018). The injection of VSLSs into the stratosphere in the form of source gases (SGs) or inorganic product gases (PGs) depends strongly on the efficiency of troposphere–stratosphere transport versus the degradation of the source gases (through photochemical loss) and product gases (through wet deposition). In particular, the question of heterogeneous release of bromine back to the gas phase, which determines the efficiency of wet deposition as a sink for Bry, is currently under discussion (e.g. Salawitch, 2006; Aschmann et al., 2011; Fernandez et al., 2014; Schmidt et al., 2016). Once brominated VSLSs have reached the stratosphere in the form of SG or PG, they participate in ozone depletion at middle and high latitudes (Braesicke et al., 2013; Yang et al., 2014; Sinnhuber and Meul, 2015). Through their relatively large impact on ozone in the lower stratosphere, they contribute −0.02 W m−2 to global radiative forcing (Hossaini et al., 2015).
The most abundant bromine-containing VSLSs are bromoform (CHBr3) and dibromomethane (CH2Br2) with atmospheric lifetime estimates ranging from 16 (50) d at the ocean surface to 29 (400) d in the tropical tropopause layer (TTL) for CHBr3 (CH2Br2) (Hossaini et al., 2012b). Both gases have potentially important source regions in tropical, subtropical, and shelf waters (e.g. Butler et al., 2007; Quack et al., 2007). The emissions of brominated VSLSs from the ocean into the atmosphere can be derived based on their concentration gradient between water and air, wind speed, sea surface temperature, and salinity (e.g. Nightingale et al., 2000; Quack and Wallace, 2003; Ziska et al., 2013). The magnitude and distribution of brominated VSLS emissions are poorly constrained given the sparse observational database of their oceanic and atmospheric concentrations (Ziska et al., 2013). Current emission inventories have been mostly derived via the top-down approach by adjusting the estimated VSLS emissions in a global atmospheric model to produce agreement of the model simulations with aircraft observations. For CHBr3, the current global top-down emissions range between 426 and 530 Gg Br yr−1 (Liang et al., 2010; Warwick et al., 2006; Ordóñez et al., 2012), while the bottom-up approach based on statistical gap filling of an observational database suggests smaller global fluxes of 164–236 Gg Br yr−1 (Ziska et al., 2013). A recent oceanic modelling study taking into account source and sink processes projects open-ocean emissions of around 72 Gg Br yr−1 in the form of CHBr3, not including the strong coastal sources (Stemmler et al., 2015). Quantitative evaluations of various emission inventories demonstrated that the performance of the individual inventories depends strongly on the region and model applied for the evaluation (Hossaini et al., 2013, 2016).
Stratospheric injection of trace gases with lifetimes of days to weeks is most efficient in regions of strong, high-reaching convective activity such as the west Pacific (e.g. Aschmann et al., 2009; Pisso et al., 2010; Marandino et al., 2013). The Asian monsoon represents another important pathway to the lower stratosphere (e.g. Randel et al., 2010; Tissier and Legras, 2016), entraining mostly South East Asian planetary boundary layer air with the potential to include emissions from the Indian Ocean and Bay of Bengal (Fiehn et al., 2017, 2018b). In both regions, the west Pacific and the Indian Ocean, these effective transport pathways may coincide with strong oceanic emissions (e.g. Ziska et al., 2013), potentially leading to anomalously large injection of brominated VSLSs. While aircraft measurements in the west Pacific have confirmed high concentrations of brominated VSLSs such as CHBr3 (Wales et al., 2018), the role of the Asian monsoon as an entrainment mechanism for VSLS is not clear due to the lack of observations in this region. Given the high variability of VSLS measurements in the TTL (Liang et al., 2010), the overall distribution and temporal short- and long-term changes are not well known. Modelling the VSLS distribution in this region depends on the magnitude and distribution of prescribed oceanic emissions, on the representation of tracer transport in the models, and on related uncertainties in both quantities (Hossaini et al., 2016). Reconciling snapshots of VSLS distributions derived from high-resolution aircraft measurements with lower spatially and temporally smoothed global modelling fields remains a challenge.
Changes in oceanic biogeochemical systems over the last decades most likely lead to changes in the marine VSLS production. However, due to the sparse data coverage and missing process understanding, it is currently not possible to quantify such long-term changes of the oceanic halocarbon production and consequences for the air–sea flux (Ziska et al., 2017). Changes in meteorological and oceanic surface parameters, which also impact the oceanic emission strength, on the other hand, have been quantified. Based on increasing sea surface temperature, salinity, and wind speed, VSLS emissions are projected to increase over the recent past (Ziska et al., 2013) and for future climate projections until 2100 (Tegtmeier et al., 2015; Ziska et al., 2017). At the same time, atmospheric transport of VSLSs is driven by changes of the atmospheric circulation. In particular, changes of tropical, high-reaching convection can be expected to have a large influence on the transport of VSLS from the ocean surface to the TTL (Aschmann et al., 2011; Hossaini et al., 2013). Long-term changes of VSLS injections into the stratosphere are difficult to predict as they are driven by various processes including changes in surface emissions, troposphere–stratosphere transport, and tropospheric chemistry (Pyle et al., 2007; Hossaini et al., 2012a).
In our study, we combine observational data sets derived during upper-TTL aircraft campaigns with Lagrangian model simulations and an observation-based VSLS emission climatology in order to analyse the spatial and temporal variability of VSLS injection into the stratosphere. Model simulations and data sets are introduced in Sect. 2. A detailed picture of the distribution of CHBr3 in the TTL (Sect. 3.1) is derived from Lagrangian transport simulations applied to a bottom-up, observation-based emission inventory. Analyses of the trajectory pathways and comparisons to aircraft observations allow us to evaluate how well we know the hotspots of CHBr3 injection (Sect. 3.2 to 3.4). We will investigate if such hotspots are mainly driven by oceanic or by atmospheric processes by analysing emission patterns and transport pathways derived from the Lagrangian simulations. We present the first estimates of the long-term changes of CHBr3 injection based on changing oceanic emissions and transport processes (Sect. 3.5). Finally, the overall contribution of CH2Br2 and CHBr3 to the stratospheric bromine loading is determined from the model simulations (Sect. 3.6) and compared to existing studies. A summary and discussion of the key results are given in Sect. 4.
2.1 Global emission climatology
The global emission scenario from Ziska et al. (2013) is a bottom-up estimate of oceanic CHBr3, CH2Br2, and CH3I fluxes. Here we focus on the two brominated compounds. Static global surface concentration maps of the two compounds were generated from atmospheric and oceanic surface ship-borne in situ measurements collected within the HalOcAt (Halocarbons in the Ocean and Atmosphere) database project (https://halocat.geomar.de, last access: May 2019). In a first step, the in situ surface measurements were classified based on physical and biogeochemical characteristics of the ocean and atmosphere important for the CH2Br2 and CHBr3 distribution and sources. In a second step, the global 1∘ × 1∘ grid was filled by extrapolating the in situ measurements within each classified region based on the ordinary least-square and robust-fit regression techniques. The method includes all in situ measurements available through the HalOcAt database at the time, regardless of season and year of the measurement. The resulting concentration maps are taken to represent climatological fields of a 23-year-long time period covering 1979 to 2013. Based on the global concentration maps, the oceanic emissions were calculated with the transfer coefficient parameterization of Nightingale et al. (2000), which was adapted to CHBr3 and CH2Br2 (Quack and Wallace, 2003). While the concentration maps do not provide any temporal variability, the emission parameterization is based on 6-hourly meteorological ERA-Interim data (Dee et al., 2011) allowing for relative emission peaks related to maxima in the horizontal wind fields and sea surface temperature. The emission inventory is available at 6-hourly, daily, and monthly temporal resolution or as a climatology product calculated as a long-term average emission field. Seasonal CHBr3 emission maps averaged over 1979–2013 are shown in the Supplement (Fig. S1).
2.2 Aircraft campaigns
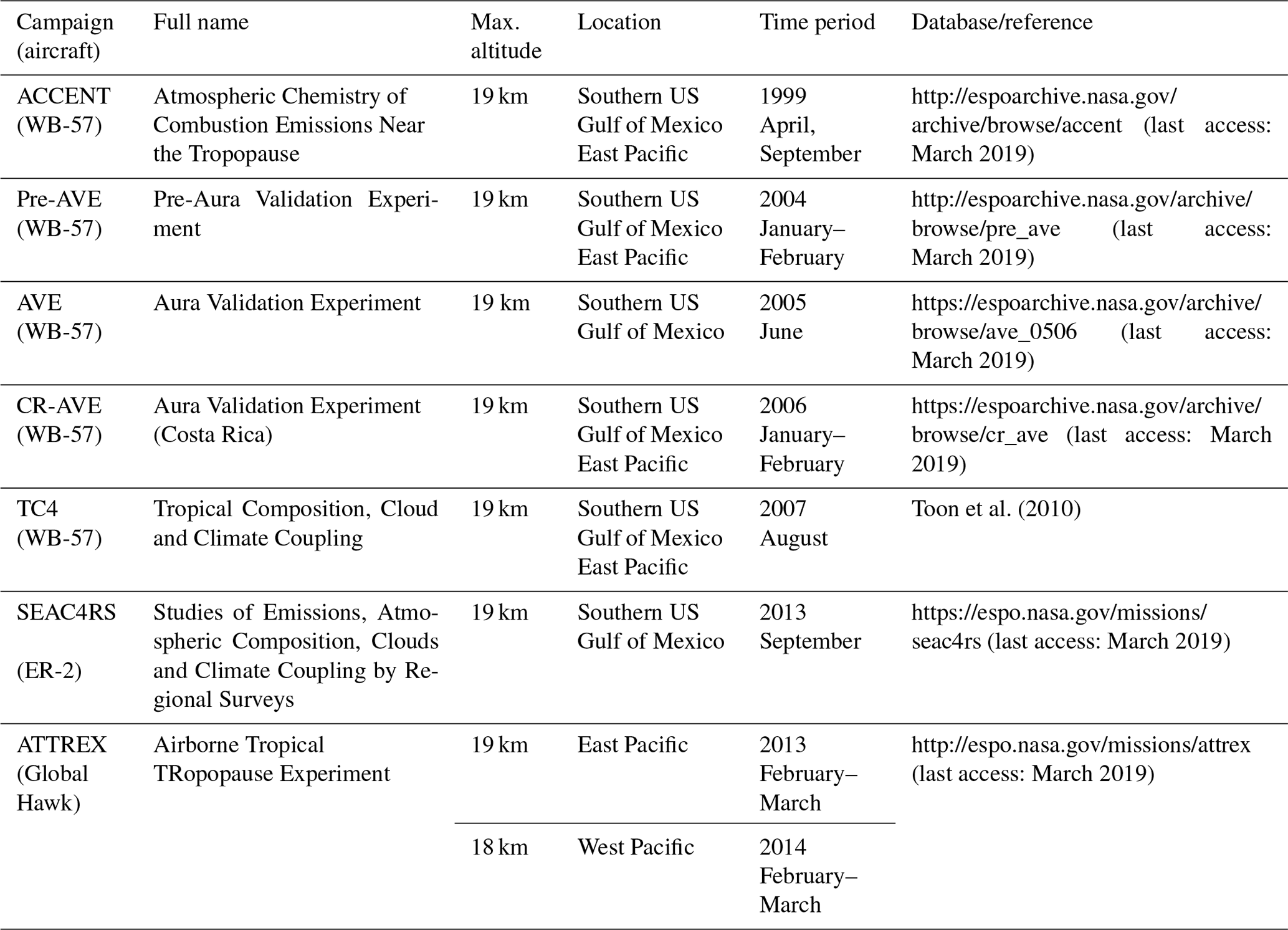
We analyse the spatial and temporal variability of CHBr3 in the TTL based on the comparison of Lagrangian transport simulations to data from aircraft campaigns. CHBr3 measurements in the upper TTL are currently available from seven aircraft campaigns. Nearly all of the campaigns took place over Central America, except for the Airborne Tropical TRopopause EXperiment (ATTREX) campaign which was in large part conducted over the Pacific. Detailed information about the aircraft missions including location and time period is presented in Table 1.
Table 1Aircraft campaigns with CHBr3 measurements used in the study.
2.3 VSLS transport modelling
We are interested in the direct contribution of CHBr3 and CH2Br2 to the stratospheric halogen loading in the form of source and product gas contributions. Therefore, the atmospheric transport of the two compounds from the oceanic surface into the upper troposphere and TTL is simulated with the FLEXPART Lagrangian particle dispersion model (Version 9.2 beta; Stohl et al., 2005, 2010). The oceanic emissions, based on the air–sea flux data from Ziska et al. (2013), prescribe the amount of CHBr3 and CH2Br2 released in the FLEXPART simulations with each air parcel trajectory. The global air–sea flux, given on a 1∘ × 1∘ grid, is used here at a monthly mean temporal resolution. For CHBr3, 90 trajectories are released per month from each grid box carrying the gas amount prescribed by the emission scenario. For the longer-lived CH2Br2, 45 trajectories are released per month. Once all brominated SGs and PGs have been removed from a trajectory through chemical decay and wet deposition, the trajectory is automatically terminated, so that the number of all active trajectories stays roughly constant (∼20 million) at all times after the initial spin-up period. The global CHBr3 simulations are run for 35 years from 1979 to 2013 with a spin-up period of 6 months in order to analyse in detail the spatial–temporal variability and long-term changes of stratospheric injection. For the longer-lived CH2Br2, the spatial–temporal variability is known to be much smaller (Hossaini et al., 2010), and runs are carried out for 3 years from 2011 to 2013 with a spin-up period of 18 months.
The transport in FLEXPART is driven by meteorological fields from the ECMWF (European Centre for Medium-Range Weather Forecasts) reanalysis model. FLEXPART includes parameterizations for moist convection (Forster et al., 2007) and turbulence in the boundary layer and free troposphere (Stohl and Thomson, 1999), dry deposition, and scavenging (Stohl et al., 2005). The runs are based on the 6-hourly fields of horizontal and vertical wind, temperature, specific humidity, convective precipitation, and large-scale precipitation from the ECMWF reanalysis product ERA-Interim (Dee et al., 2011) given at a horizontal resolution of 1∘ × 1∘ on 60 model levels. A preprocessor retrieves the meteorological fields from the ECMWF archive, including the vertical wind, which is calculated in hybrid coordinates mass-consistently from spectral data. FLEXPART has been validated based on comparisons with measurement data from three large-scale tracer experiments (Stohl et al., 1998) and with results from intercontinental air pollution transport studies (e.g. Forster et al., 2001; Stohl and Trickl, 1999). Previous FLEXPART studies using a similar model setup as applied here have shown a very good agreement between diagnosed and observed VSLS profiles (e.g. Tegtmeier et al., 2013; Fuhlbrügge et al., 2016).
FLEXPART includes the simulation of chemical decay by reducing the tracer mass carried by each air parcel corresponding to its prescribed chemical lifetime. We set the atmospheric lifetime of CHBr3 (CH2Br2) to an altitude-dependent lifetime profile ranging from 16 (50) d at the ocean surface to 29 (400) d in the TTL (Hossaini et al., 2012b). The lifetime profiles were derived from simulations of the chemical tropospheric loss processes of CHBr3 and CH2Br2 with the chemical transport model TOMCAT (Chipperfield, 2006). Previously, profiles from TOMCAT have been shown to agree well with aircraft observations in the tropical troposphere (Hossaini et al., 2012b).
The bromine resulting from the photochemical loss of CHBr3 and CH2Br2, based on prescribed loss terms, contributes to the inorganic product gases. In the FLEXPART simulations, these product gases are grouped together as Bry and transported together with the VSLS source gases along the trajectory. Thus, we assume instantaneous conversion between organic intermediate product gases and Bry, which has been shown to be reasonable by Hossaini et al. (2010). Bry can be removed effectively from the troposphere through wet scavenging by rain or ice (Yang et al., 2005). FLEXPART includes in-cloud as well as below-cloud scavenging, which is initiated if the relative humidity as calculated from meteorological input data exceeds 80 % and the precipitation rate is larger than zero. In FLEXPART, the cloud scavenging ratio is used to model washout of soluble species. The ratio is calculated within FLEXPART with the help of the effective Henry's law coefficient, Heff, which describes the physical solubility of a species as well as the effects of dissociation. Among the members of the Bry family, HBr and HOBr can be washed out while the remaining species Br, BrO, BrONO2, and Br2 are not soluble. HBr has a very large acidity dissociation constant resulting in an effective Henry's law coefficient of 7.1×1013 M atm−1 for T=298 K and pH = 5 (Yang et al., 2005). While HBr provides the main pathway for wet removal of inorganic Bry, HOBr is also soluble due to physical solubility, but not due to dissociation (Frenzel et al., 1998) with M atm−1. In order to determine which fractions of Bry are in the form of HBr and HOBr, we apply the Bry partitioning derived from p-TOMCAT simulations (Yang et al., 2010).
Based on analysed wind fields together with complex chemical schemes, p-TOMCAT simulates the tracer distribution in the troposphere and lower stratosphere including gaseous phase bromine chemistry. The three-dimensional Bry field from p-TOMCAT and its partitioning into HOBr, HBr, Br, BrO, BrONO2, and Br2 are given at a time step of 30 min. As the partitioning of the Bry field varies strongly with location and time, we apply it in a first step to every air parcel according to its location each time before the wet deposition is initiated. In a second step, wet deposition is calculated individually for each inorganic bromine species based on its solubility specified by the effective Henry's law coefficient, as described above. Once wet deposition is initiated the Bry fraction determined to be washed out is removed completely.
Dissolved inorganic bromine can be released back to the gas phase by heterogeneous chemical reactions (Abbatt, 2003; Salawitch, 2006), extending the tropospheric lifetime of Bry by altering the efficiency of wet deposition (von Glasow et al., 2004). The heterogeneous reactions on aerosols which reactivate bromine radicals from the reservoir species (Yang et al., 2005, 2010) are included in the chemical scheme of p-TOMCAT. This release of bromine back to the gas phase results in elevated BrO∕Bry ratios (Yang et al., 2010), and thus a lower fraction of Bry is subject to wet deposition compared to a scenario without the heterogeneous chemical reactions. As we use the p-TOMCAT Bry partitioning for our trajectory simulations, these aerosol effects have indirectly been taken into account when simulating the wet removal of Bry. Uncertainties in the modelled wet deposition arise from the parameterization of solubility via the effective Henry's law coefficient and from uncertainties in the Bry partitioning caused by errors in the aerosol loading and in the mechanism used for heterogeneous reactions. Clouds and aerosols within p-TOMCAT are not matched with those in FLEXPART, which might lead to an additional error source.
For the analysis of the spatial and temporal variability of CHBr3 in the TTL from FLEXPART simulations and aircraft observations in Sect. 3.1 to 3.5, we use mixing ratios at 17 km (approximate cold point) and mixing ratios averaged over 16–18 km (upper part of the TTL). In order to derive the amount of VSLS source and product gases entrained into the stratosphere from the model simulations in Sect. 3.6, we explicitly calculate the cold point along each trajectory based on the ERA-Interim meteorological fields as stratospheric entrainment point. The derived estimates of stratospheric VSLS entrainment depend on the meteorological input data sets and on various FLEXPART model parameters, such as the convective parameterization. The accurate representation of convection has been validated with tracer experiments and 222Rn measurements (Forster et al., 2007). The application of transport timescales based on vertical heating rates instead of vertical wind fields in the TTL between 15 and 17 km results in only minor differences of VSLS entrainment (Tegtmeier et al., 2012).
3.1 CHBr3 in the TTL
Figure 1a shows the long-term annual mean CHBr3 distribution at 17 km as derived from the Lagrangian transport calculations driven by monthly mean oceanic emission fields for the time period 1979–2013. Clearly, CHBr3 has a very pronounced spatial variability due to its short lifetime. The largest CHBr3 mixing ratios of up to 0.4 to 0.5 ppt can be found over (1) Central America, (2) the Maritime Continent and the tropical west Pacific, and (3) the tropical Indian Ocean (all regions are highlighted by black squares in Fig. 1a labelled from 1 to 3). Other tropical regions with only little convective uplift show smaller mixing ratios, mostly between 0.1 and 0.2 ppt.
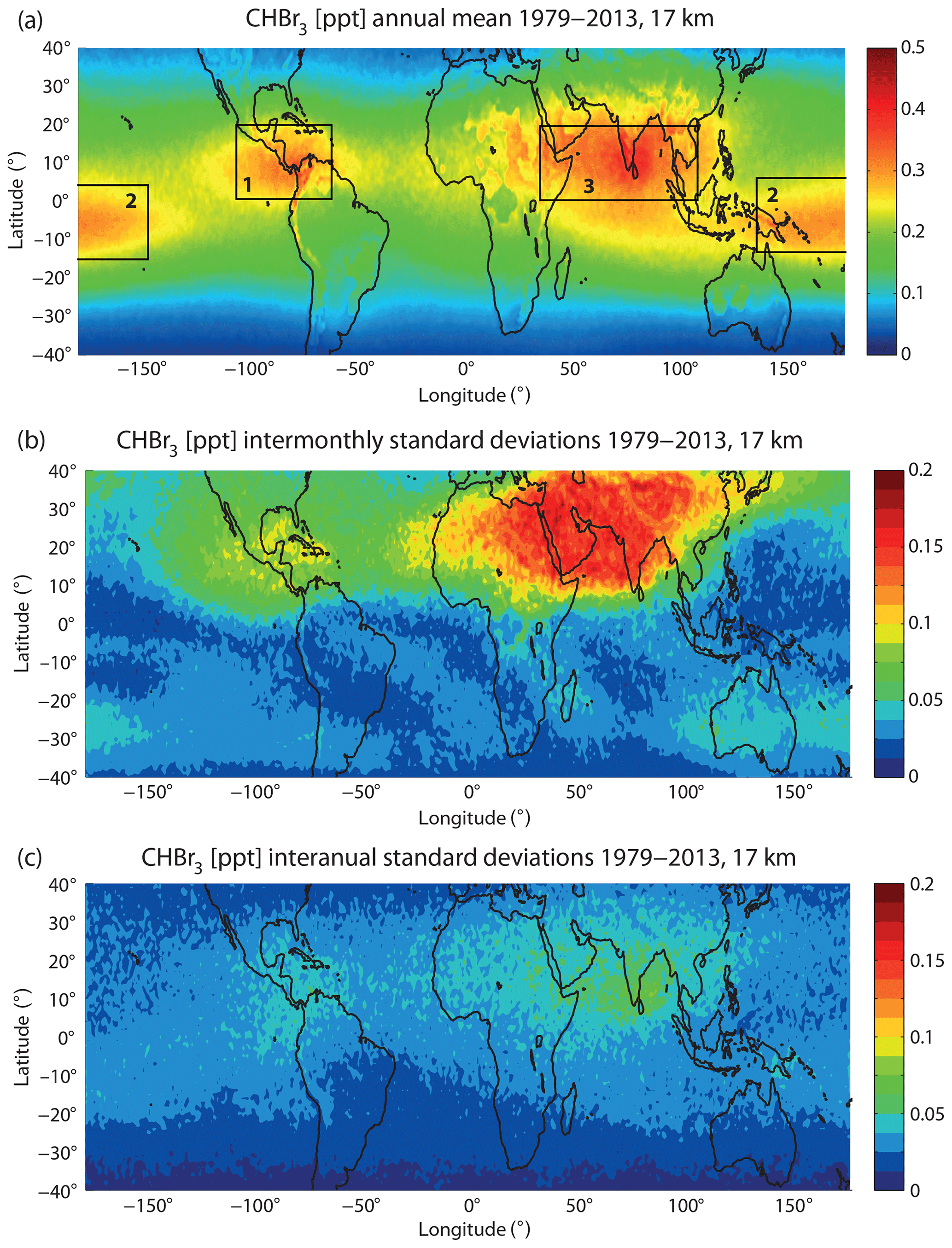
Figure 1Modelled annual mean distribution of CHBr3 at 17 km for 1979–2013 (a) and the inter-monthly (b) and inter-annual (c) variations given by the standard deviations over all monthly, multi-annual mean, and annual mean values, respectively. Rectangles stand for regions of maximum CHBr3 mixing ratios and will be discussed in detail in Sect. 3.2 to 3.4.
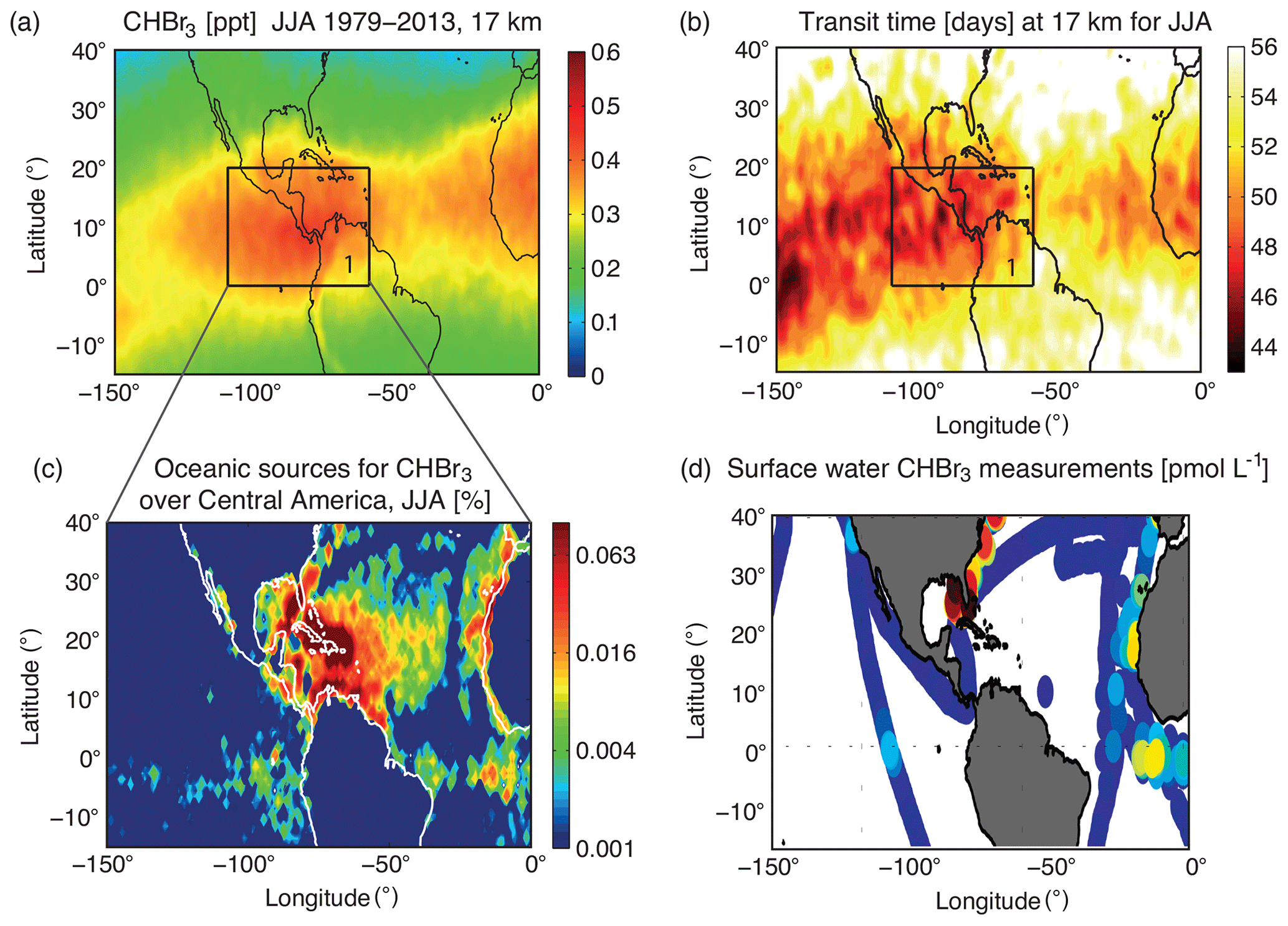
Figure 2Modelled distribution of CHBr3 at 17 km for JJA, 1979–2013 (a), transit time of air masses from the ocean surface to the TTL (b), oceanic source regions for CHBr3 (c), and measurements of oceanic CHBr3 concentrations from the HalOcAt database used for Ziska et al. (2013) (d). The oceanic source regions in (c) are colour coded according to their contribution (% per 1∘ × 1∘ grid box) to the amount of CHBr3 at 17 km in the black box over Central America (highlighted in a and b).
Entrainment of CHBr3 into the stratosphere also shows a large temporal variability. The seasonal variability is given here by the standard deviation over all monthly, multi-annual mean values (Fig. 1b). The by far most pronounced variability is found in the region of the Asian monsoon anticyclone, which is characterized by a strong seasonality of vertical transport processes (Randel et al., 2010). Furthermore, the distribution of CHBr3 at the cold point over Central America shows some seasonal variations, however, of smaller magnitude. The Maritime Continent and tropical west Pacific have only a very weak seasonal cycle. Overall, the seasonal variations are more pronounced in the Northern Hemisphere (NH) tropics and quite low in the Southern Hemisphere (SH) tropics. Seasonal CHBr3 entrainment averaged over 1979–2013 is shown in the Supplement (Fig. S2).
Inter-annual variations are given in the form of the standard deviation over all annual mean CHBr3 mixing ratios at 17 km (Fig. 1c). In comparison to the seasonal variability, the inter-annual variability is relatively small in the NH tropics but is of similar magnitude in the SH tropics. Drivers of the seasonal and inter-annual variability will be discussed in the following sections. We will analyse the three regions with maximum CHBr3 entrainment identified above and investigate the relative importance of emissions and transport processes for the overall distribution and seasonality of stratospheric injection.
3.2 Central America
CHBr3 in the TTL, on its way from the ocean surface to the stratosphere, shows a pronounced maximum over Central America between 0–20∘ N and 60–110∘ W (black square in Figs. 1a and 2a). This maximum is present all year, but most pronounced during NH summer and autumn. In the following, we will use the simulations for June–July–August to address the question of whether this maximum arises from very strong oceanic sources or from strong convective transport. The impact of transport on the CHBr3 distribution in the upper TTL is analysed by estimating the time air masses need from the ocean surface to 17 km based on the FLEXPART model simulations. The transport time of each trajectory is assigned to the location where the trajectory reaches 17 km. A map of the “ocean surface–17 km transit times” is derived by averaging over all trajectories on a 1∘ × 1∘ grid. The tropical annual mean transit time is around 55 d with variations between 45 and 70 d (not shown here). Transit times over Central America for the June–July–August season are relatively short with values around 48 d (Fig. 2b). However, the transit times over the east and central Pacific are similar or even shorter, suggesting that the vertical transport in this region is as efficient as over Central America. Therefore, atmospheric transport timescales alone cannot explain the CHBr3 maximum over Central America.
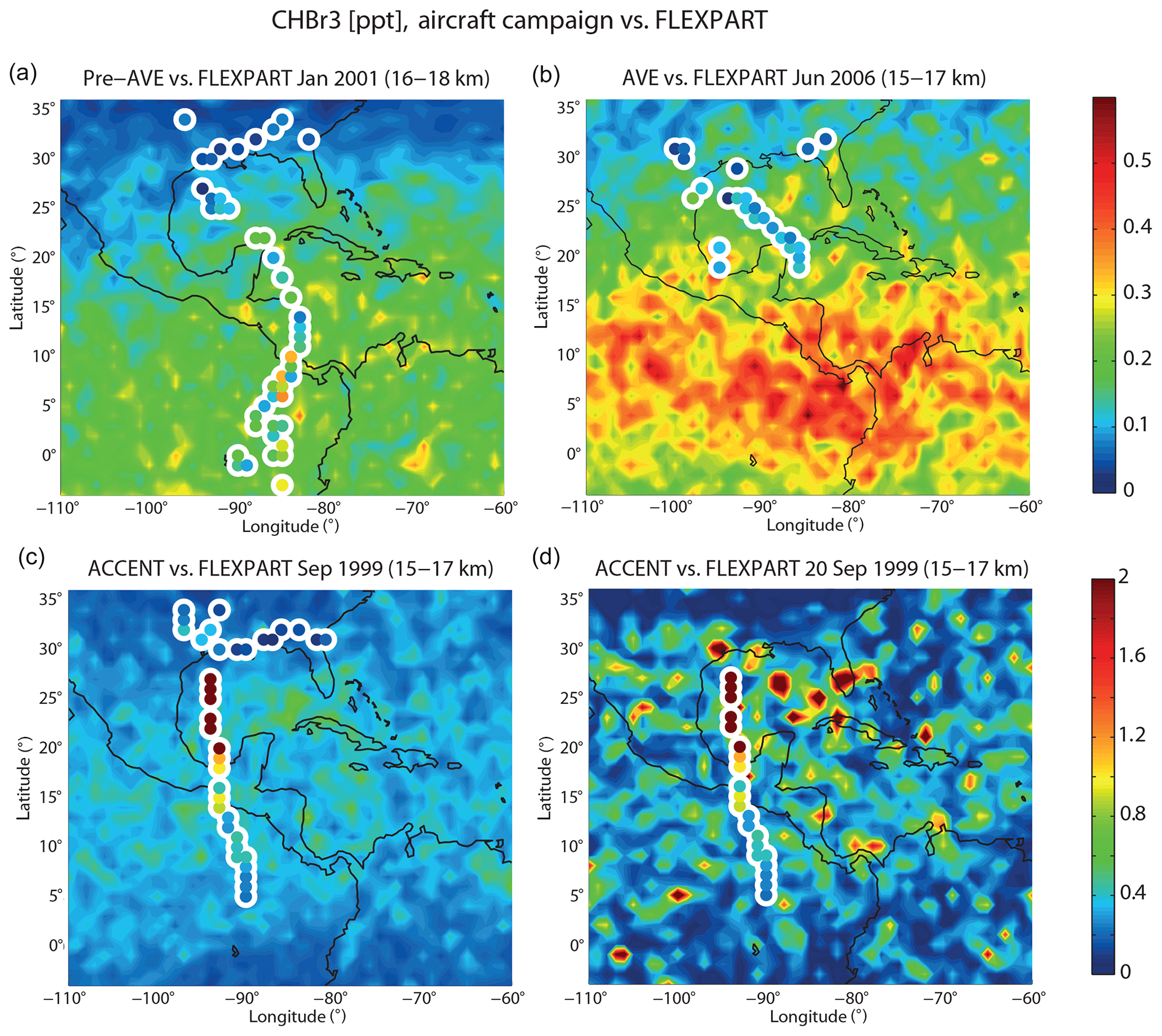
Figure 3Modelled distribution of CHBr3 in the upper TTL from FLEXPART (background colouring) in comparison with aircraft campaign measurements (coloured symbols with white edges). In (a), (b), and (c), all individual measurements from the respective campaign and the model mean over the same time period are shown. Only in (d) is one individual flight (ACCENT flight from 20 September 1999) shown together with FLEXPART daily mean values to illustrate the large spatial variability including maximum values ≥2 ppt.
In addition to the transit time, we analyse the oceanic sources of CHBr3 over Central America. Each trajectory reaching the TTL over Central America (black square in Fig. 2a) contributes a certain amount of CHBr3 to this local maximum by carrying its prescribed oceanic emission (Ziska et al., 2013) from the surface to the cold point. The relative contribution (%) of each trajectory is assigned to its oceanic release point, thus quantifying which ocean region contributes the largest amounts of CHBr3 to the local maximum in the TTL. The relative contributions averaged over 1∘ × 1∘ grid cells (Fig. 2c) demonstrate that the largest sources stem from the Gulf of Mexico, the Caribbean Sea, and the western North Atlantic. Some smaller contributions come from the west coast of North Africa and from the equatorial Atlantic. The co-occurrence of strong sources and the relatively short transport timescales over the Caribbean Sea and Central America mainly cause the local CHBr3 maximum in the Central American TTL. While transport timescales are also short (or even shorter) in the eastern Pacific, oceanic emissions are very small there, and vice versa more pronounced emissions over the Atlantic and along the coast of Africa do not cause a global maximum due to longer transport timescales.
The regional oceanic measurements in surface water, which were used to derive the extrapolated concentration and emission maps (Ziska et al., 2013), are given in Fig. 2d. The available data show, in particular, high oceanic CHBr3 concentrations at the Florida coastline and in the eastern part of the Gulf of Mexico. A reasonable number of measurements with a distinctive distribution are available in this region, supporting the extrapolated climatological source distribution, which leads to the CHBr3 maximum in the TTL over Central America discussed above.
Over the last decades, the atmospheric distribution of CHBr3 over Central America has been investigated by a number of different aircraft campaigns. We will use available upper air measurements to evaluate the distribution and variability of the model-derived CHBr3 fields. Details of the aircraft campaigns are given in Table 1. We show the spatial CHBr3 distribution in the TTL as observed during three different campaigns in comparison to the model simulations (Fig. 3). The altitude ranges in the upper TTL have been chosen so that each comparison includes a maximum number of observational data. While for the aircraft campaigns individual measurements are shown at the measurement locations, the model fields are averaged over the duration of the respective campaign. This method allows us to evaluate the spatial distribution of measured and modelled CHBr3 fields, but it has the disadvantage of comparing in situ data with temporally averaged fields. We will discuss how this can impact the comparison and how the temporal variability can be taken into account.
For the Pre-AVE campaign during NH winter, CHBr3 in the upper TTL (16–18 km) shows a latitudinal gradient with small values of 0–0.1 ppt in the northern subtropics and with higher values of up to 0.3–0.4 ppt around the Equator. The same gradient is also evident from the model simulation, resulting in an overall good agreement. Similarly, for the AVE campaign during NH summer, both the observations and the model results show a latitudinal gradient with increasing values towards lower latitudes. However, here the overall agreement is poor, since the model results are on average 50 % larger than the measurements.
Finally, for the ACCENT campaign during NH autumn, the observations reveal extremely high CHBr3 (up to 2 ppt) between 30 and 20∘ N. While CHBr3 is decreasing north and south of this area towards the range of 0.5–1 ppt, the values are still very high when compared to other campaigns over Central America. FLEXPART results, averaged over the time period of the ACCENT campaign (September 1999), show the largest monthly mean CHBr3 values of around 0.7 ppt, which are substantially smaller than the observations of 2 ppt. However, the model results look quite different and show large spatial inhomogeneities when evaluated at a daily mean resolution. Maximum model values are much higher for the daily resolution and in some locations, very close to the flight track, of similar size as the observations (around 2 ppt). The spotty features in the model simulations are a result of the high oceanic sources directly underneath interacting with localized convective transport. The latter brings localized air masses with very high CHBr3 mixing ratios from the boundary layer into the 15–17 km layer. The differences between monthly and daily mean model values make clear that CHBr3 model–measurement comparisons may be obscured by the high variability of the field. Given this high variability and the existing uncertainties in the diagnosed oceanic sources and atmospheric transport processes, it is very difficult for a model to predict the correct in situ values at a given time and measurement position. Nevertheless, if the large-scale emissions and transport fields are correct, spatial and temporal averaging of the model results can be expected to produce realistic mean VSLS fields and to improve the agreement with observations. Only in cases where rare events have been observed will averaging the CHBr3 fields not necessarily lead to a better agreement with the measurements, as demonstrated above for the ACCENT campaign. Consequently, it is important to include estimates of the spatial and temporal variability of the CHBr3 field in all comparisons.
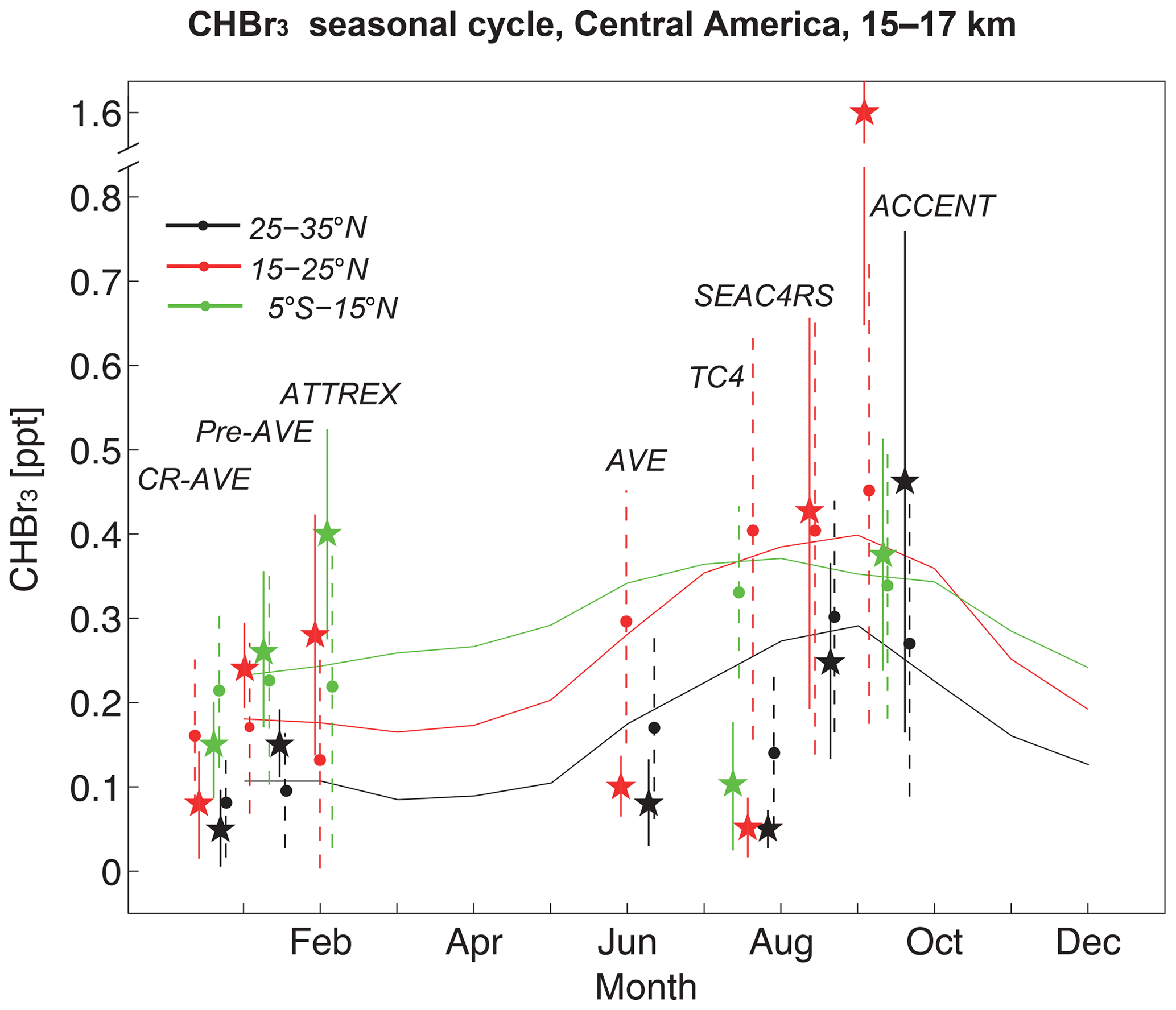
Figure 4Seasonal cycle of CHBr3 in the upper TTL (15–17 km) over Central America from FLEXPART simulations (solid lines) averaged over 110–80∘ W and 5∘ S–15∘ N (green), 15–25∘ N (red), and 25–35∘ N (black) for 1999–2013. In addition, aircraft measurements (stars) and coincident FLEXPART values (filled circles) are shown averaged over the same latitude bins and corresponding to the respective year of the campaign. Temporal and spatial variability of average measurements (solid vertical lines) and coincident model values (dashed vertical lines) is shown in the form of the 1-sigma standard deviation over all values in the respective bin.
A summary of the CHBr3 model results compared to aircraft campaigns in the Central American region, taking into account spatial and temporal variability, is provided in Fig. 4. Here, we compare measurements averaged over different parts of the flight tracks (split by latitude) with FLEXPART coincidences averaged over the same latitudinal bins. The variability of the CHBr3 distribution from observations and coincident model values is given by the standard deviation over all values in the respective region. In addition, the FLEXPART seasonal cycle averaged over 110–80∘ W, the main longitudinal extent of the aircraft campaigns, and the entire campaign time period (1999–2013), is shown. The comparison of the three campaigns during NH winter shows an overall good agreement. For some latitude bins, the modelled mean values agree very well with the observations (e.g. Pre-AVE for 5∘ S–15∘ N); for other regions, differences of the mean values can be up to 50 %–100 %. However, all observational mean values are within the standard deviations of the modelled field, indicating good agreement of model and measurements.
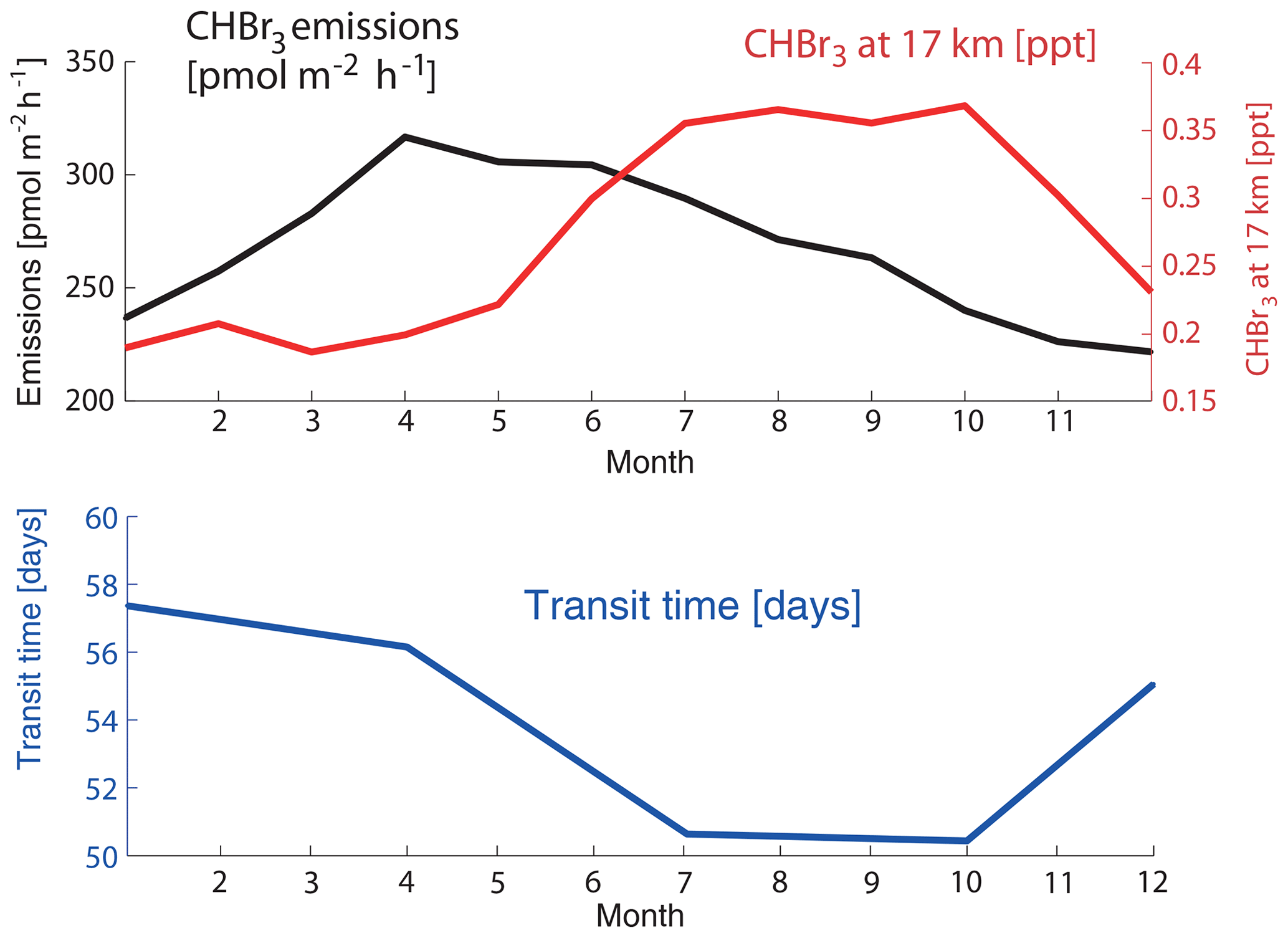
Figure 5Seasonal cycles of CHBr3 at 17 km over Central America (black square in Fig. 2a) from FLEXPART simulations (red line), oceanic CHBr3 emissions averaged over the respective source region (black line), and the “surface–17 km” mean transit time (blue line) are shown.
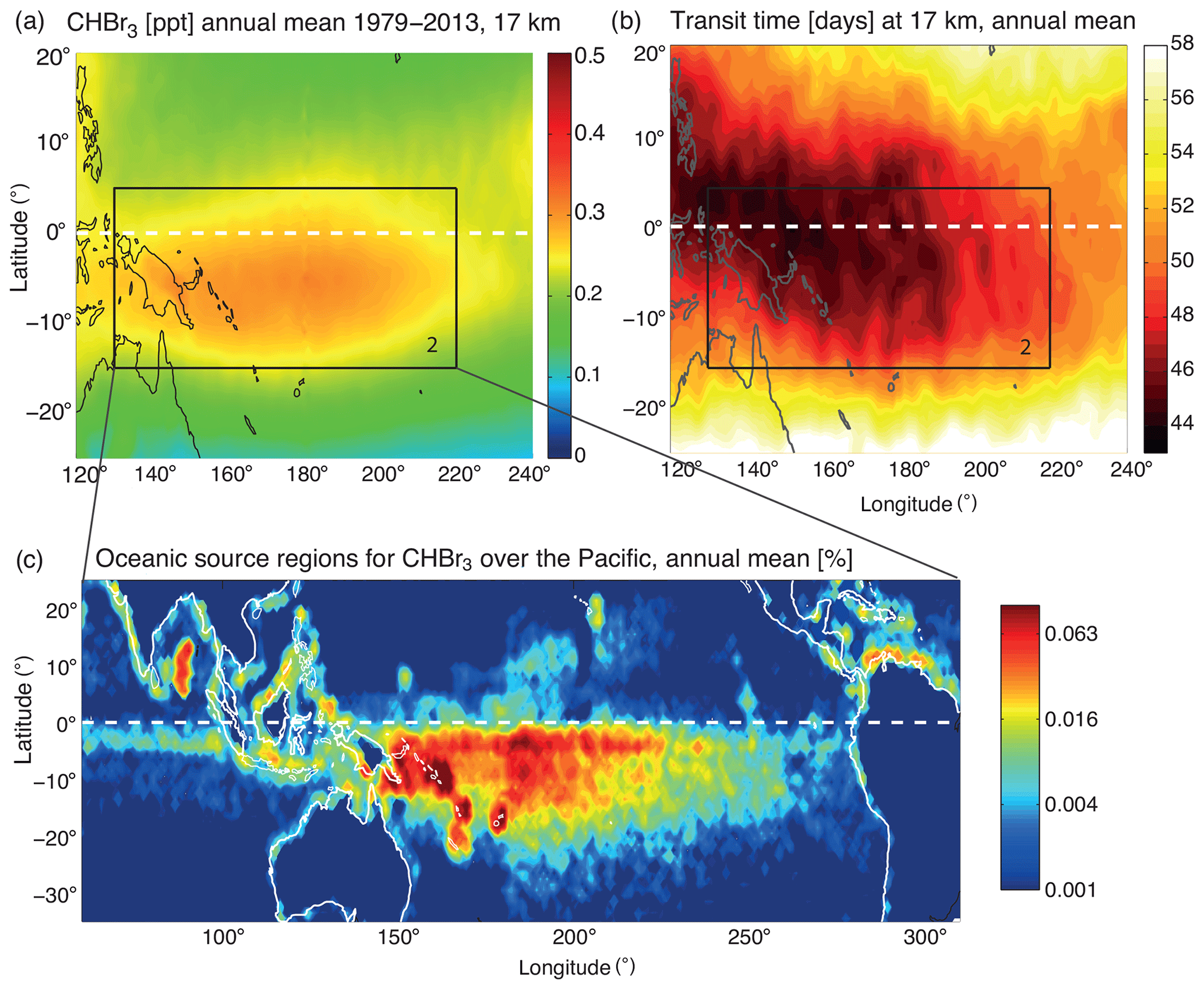
Figure 6Modelled distribution of CHBr3 at 17 km, annual mean 1979–2013 (a), transit time of air masses from the ocean surface to the TTL (b), and oceanic source regions for CHBr3 at 17 km (c). Oceanic source regions are colour coded according to their contribution (% per 1∘ × 1∘ grid box) to the amount of CHBr3 at 17 km in the black box over the west Pacific (highlighted in a and b).
For the campaigns during NH summer, mean differences are in general larger than during NH winter. At the same time, the temporal and spatial variability of the simulated and observed CHBr3 distribution is also larger so that most observations agree with the coincident model values within their uncertainties. The large differences between the individual campaigns during NH summer confirm the increased variability suggested by the model results. For two of the campaigns (AVE and TC4), FLEXPART overestimates the CHBr3 values during this time of the year, while for the other two campaigns (SEAC4RS and ACCENT), the observations and modelled values agree relatively well except for one outlier. Particularly high CHBr3 exists for the 15–25∘ N region, observed during the ACCENT campaign at the top altitude of a plume extending from 14 to 16 km near Houston, Texas. This value is larger than the model mean, although observational and model value uncertainties slightly overlap. In total, observations and model agree reasonably well with a larger variability during the NH summer and early autumn period. For this time of the year, the model also suggests a seasonal CHBr3 maximum which is confirmed by measurements from SEAC4RS and ACCENT, but not by the AVE and TC4 campaigns.
CHBr3 in the upper TTL over Central America shows pronounced seasonal variations as revealed by the comparisons to aircraft campaigns in Fig. 4. The CHBr3 seasonal cycle at 17 km shows a maximum from July to October (∼0.37 ppt) and a minimum from January to April (∼0.17 ppt) (Fig. 5a). Such seasonal variations can be caused by variations in the oceanic emissions or the atmospheric transport times. First, we analyse the seasonal cycle of CHBr3 emissions, averaged over the source region identified earlier, which show peak emissions from April to June of up to 320 pmol m−2 h−1. This peak in surface emissions in late spring–early summer is consistent with a peak in the TTL around 2 months later, as the mean transit time from the surface to 17 km in this region is about 55 d. Second, we analyse the seasonal cycle of the transit time and find a minimum from July to October, which is also consistent with the highest CHBr3 values in the TTL during the same time period. While the amplitude of the seasonal cycle in CHBr3 in the TTL is around 74 %, seasonal variation in the emissions and the transit time are only 36 % and 15 %, respectively. However, the amplitude in transit time does not directly translate into the amplitude in CHBr3 in the TTL, given the logarithmic nature of the atmospheric lifetime of chemical compounds. Overall, the interaction of both processes, oceanic emissions and atmospheric transport, causes the pronounced seasonal cycle of CHBr3 over Central America.
3.3 Maritime Continent and tropical west Pacific
CHBr3 in the TTL shows a pronounced maximum over the Maritime Continent and tropical west Pacific between 15∘ S–5∘ N and 130–220∘ E (black square in Figs. 1a and 6a). An important characteristic of this CHBr3 maximum (referred to as the west Pacific maximum hereinafter) is that the high values are not distributed symmetrically across the Equator but are shifted southwards. The maximum is present all year with no pronounced seasonal cycle (see Fig. 1b). In the following, we will use annual mean results to investigate if the high values arise from very strong oceanic sources or from strong convective transport. The transit time shows the smallest values of around 45 d in the west Pacific and over the Maritime Continent (Fig. 6b). The most important deviation from the CHBr3 distribution at 17 km is that over the west Pacific the shortest timescales and thus most efficient transport are not centred in the SH, but they are distributed symmetrically across the Equator.
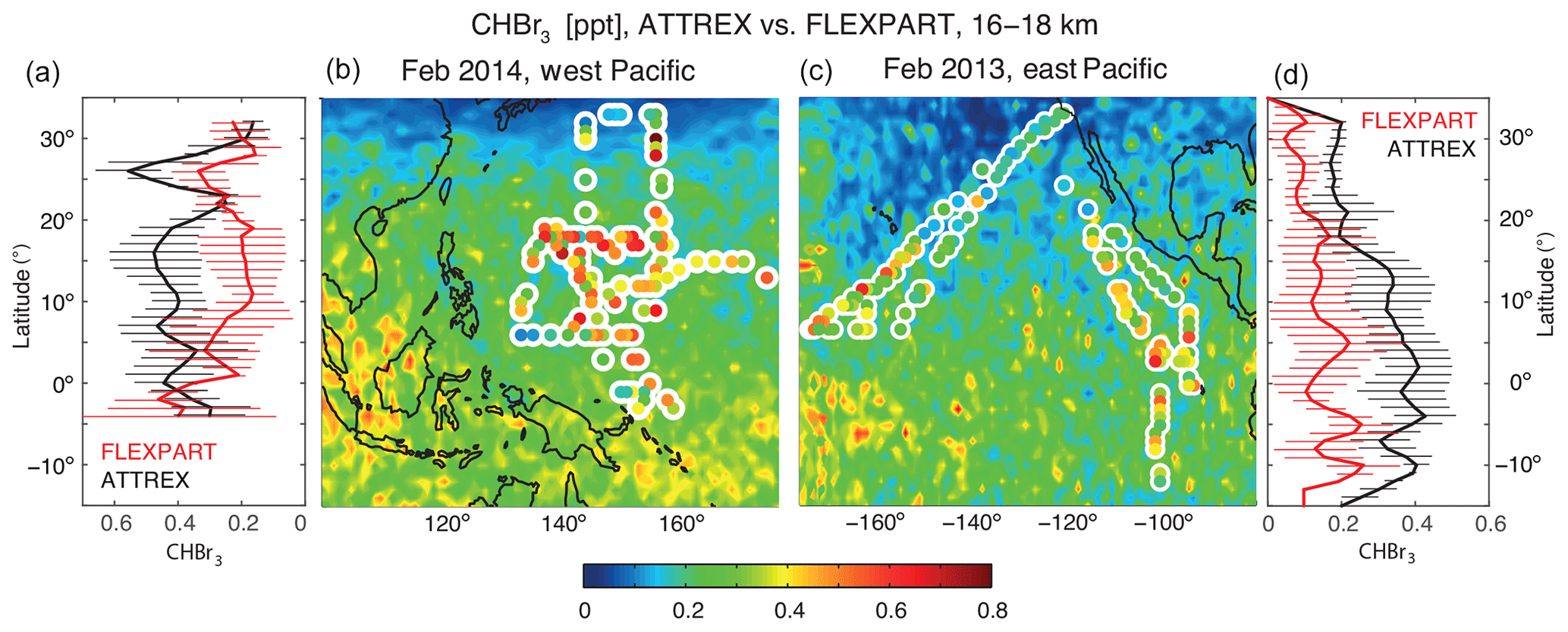
Figure 7Modelled distribution of CHBr3 in the uppermost TTL from FLEXPART (background colouring) in comparison with ATTREX aircraft campaign measurements (coloured symbols with white edges) is given in (b) and (c). Zonal means of coincident model–measurement comparisons are given in (a) for FLEXPART and the ATTREX campaign in February–March 2014 in the west Pacific and in (d) for FLEXPART and the ATTREX campaign in February–March 2013 in the east Pacific. Temporal and spatial variability of measurements and coincident model values is shown in form of the 1σ standard deviations over all values in the respective zonal bin (horizontal lines).
Oceanic sources for CHBr3 in the west Pacific upper TTL (black square in Fig. 6a) stem mostly from the Pacific Ocean, the Maritime Continent, and also to a smaller degree Central America (Fig. 6c). The trajectory analysis clearly shows that the largest contribution comes from the west Pacific south of the Equator, while the oceanic contributions north of the Equator are lower. This pattern is directly related to the emission inventory used in this study (Ziska et al., 2013), which suggests overall stronger emissions in the southern Pacific Ocean (see Fig. S1 in the Supplement). However, available open-ocean surface measurements in both the NH and SH Pacific Ocean were sparse during the time of the construction of the inventory and mostly based on the TransBrom Sonne campaign (Krüger and Quack, 2013). The latitudinal gradient of the emission inventory with stronger emissions in the SH is based on the in situ measurements along one cruise track from Japan to Australia during October 2009 and may not be representative for other seasons and other west Pacific regions. Future ship campaigns are necessary to confirm or improve the existing emission inventory.
Pacific aircraft campaigns are used to further analyse the hemispheric differences of the diagnosed CHBr3 distribution. ATTREX measurements in the west Pacific in 2014 and in the east Pacific in 2013 are compared to FLEXPART simulations in Fig. 7. In both regions, the comparison reveals a reasonably good agreement with increasing CHBr3 values towards lower latitudes. In the west Pacific, measurements and coincident model values agree best south of 10∘ N, while north of this the model underestimates observations by up to 0.3 ppt. In the east Pacific, model values and measurements are closer in the NH and agree mostly within their error bars. South of the Equator, however, measurements are constantly larger with differences of up to 0.3 ppt. In total, the modelled CHBr3 entrainment over the Pacific is too small when compared to measurements, which could be due to an underestimation of the oceanic emissions in this region.
3.4 Tropical Indian Ocean
Annual mean CHBr3 in the uppermost TTL shows a pronounced maximum over India, the Bay of Bengal, and the Arabian Sea between 2–22∘ N and 35–110∘ E (Fig. 1a, referred to as the Indian Ocean maximum hereinafter). The simulations diagnose the globally highest TTL CHBr3 values of up to 0.5 ppt in the long-term mean over the southern tip of India. At the same time, the intermonthly standard deviation is very high over this region (Fig. 1b) due to pronounced seasonal variations. During NH summer (June–July–August), high CHBr3 values of around 0.6 ppt are found over a large region stretching from South East Asia all the way to northeast Africa between 10 and 25∘ N. During SH summer (December–January–February), smaller maximum values of around 0.4 ppt CHBr3 are diagnosed south of India over the Indian Ocean between 5∘ S and 10∘ N (Fig. 8).
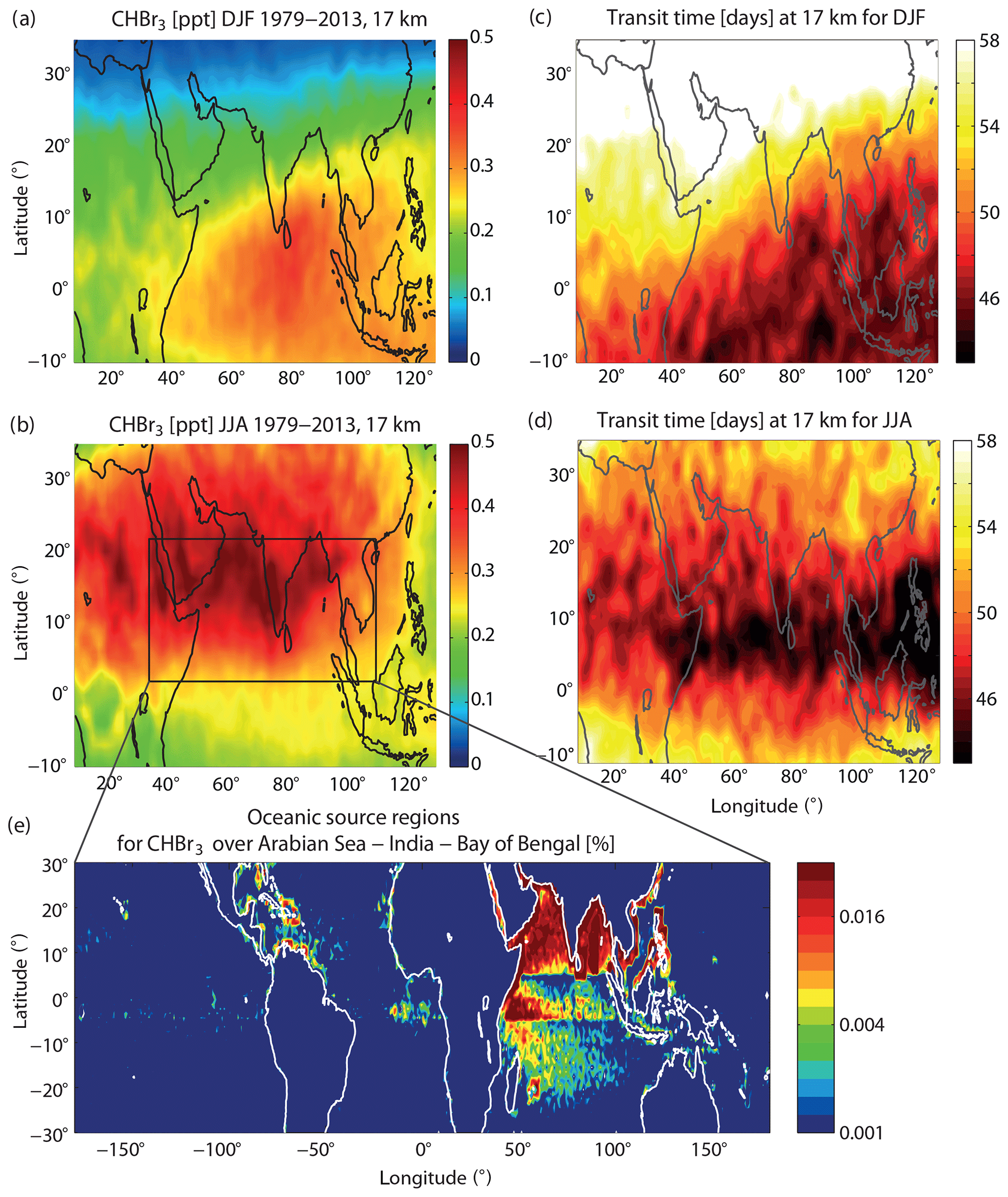
Figure 8Modelled distribution of CHBr3 at 17 km for DJF and JJA 1979–2013 (a, b). Transit time of air masses from the ocean surface to the TTL for DJF and JJA (c, d). Oceanic source regions colour coded according to their contribution to CHBr3 at 17 km over Arabian Sea, India, and Bay of Bengal (black box in b) given in percent per 1∘ × 1∘ grid box (e).
In order to evaluate the transport efficiency for oceanic short-lived trace gases in this region, the transit time is calculated from the trajectory analysis for the NH and SH summer seasons. During NH winter, transit times from the surface to the TTL show a very similar pattern as CHBr3 in the TTL, with the shortest transit times of around 45 d over the Indian Ocean coinciding with the largest CHBr3 abundance. During NH summer, on the other hand, the transit times minimize not in the region of maximum CHBr3 abundance, but instead south of this region where air masses can reach the TTL within 43 d. Between 10 and 25∘ N, the transport is still fast and the transit of short-lived species from their ocean sources will take around 48 d. Overall the transit time is similar to values found for the west Pacific and cannot solely account for the simulated maximum CHBr3 values.
CHBr3 contributing to the Indian Ocean TTL maximum mostly stems from the Bay of Bengal, the Arabian Sea, the equatorial region of the Indian Ocean, and the coastlines of South East Asian countries like China. Compared to the oceanic contributions identified for the Central America and west Pacific maxima, sources for the Indian Ocean CHBr3 maxima show a large regional extent including coastal and open-ocean emissions from 20∘ S to 30∘ N. Given that oceanic emissions from large parts of the Indian Ocean and adjacent coastal areas can be transported into the Asian monsoon region (Fiehn et al., 2017), the CHBr3 maxima can be explained by the strong oceanic emissions in this region combined with efficient boundary layer–TTL transport.
The global maximum of CHBr3 over India, Bay of Bengal, and the Arabian Sea is also subject to the largest uncertainties when compared to the other maxima found in our model simulations. For the construction of the emission inventory from Ziska et al. (2013), only one data set was available for the Indian Ocean (Yamamoto et al., 2001). The data set is based on measurements at seven stations in the open-ocean waters of the Bay of Bengal and reveals relatively high CHBr3 values between 8 and 15 ng L−1. Given the great distance of the sampling points from the coasts, the authors hypothesized that planktonic production is the most probable source for this high CHBr3 abundance. Independent measurements from the OASIS campaign in 2014 confirm the subtropical and tropical west Indian Ocean as a strong source for CHBr3 to the atmosphere, although open-ocean surface concentrations were overall lower with maximum values of 8 ng L−1 (Fiehn et al., 2017). A recent update of the Ziska bottom-up CHBr3 emission climatology (Fiehn et al., 2018b) suggests enhanced emissions in the tropical Indian Ocean, which would lead to even higher stratospheric entrainment in this region. While the high values from Yamamoto et al. (2001) were used locally for the emission climatology, the rest of the tropical Indian Ocean was filled by applying open-ocean data from the tropical Atlantic and Pacific. Consequently, the emission scenario for the Indian Ocean has large uncertainties, and further VSLS measurements are required to confirm or improve our estimates of the Indian Ocean as the region of strongest CHBr3 entrainment into the stratosphere.
3.5 Inter-annual and long-term changes
Long-term changes of tropical mean (30∘ N–30∘ S) CHBr3 mixing ratios at 17 km show a weak but significant trend of 0.017±0.012 ppt Br per decade, corresponding to a 10 % increase in CHBr3 over the whole time period (1979–2013). Regionally, the long-term changes are more pronounced and FLEXPART simulations suggest decreasing or increasing CHBr3 in the TTL depending on the location (Fig. 9). Over South America, Australia, and the central–east Pacific, the trend is not significant given the relatively small trend values compared to the inter-annual variability found here. For all other regions, CHBr3 shows a significant positive trend of 2 %–10 % per decade. CHBr3 over the Indian Ocean and Maritime Continent is highlighted in Fig. 9c as the region with the maximum trend (0.04 ppt Br per decade), mostly driven by the steep changes related to El Niño–Southern Oscillation (ENSO) over the time period 2000–2013 (Fiehn et al., 2018a). CHBr3 over the east Pacific is highlighted in Fig. 9b as an example of a negative but not significant trend (−0.017 ppt Br per decade).
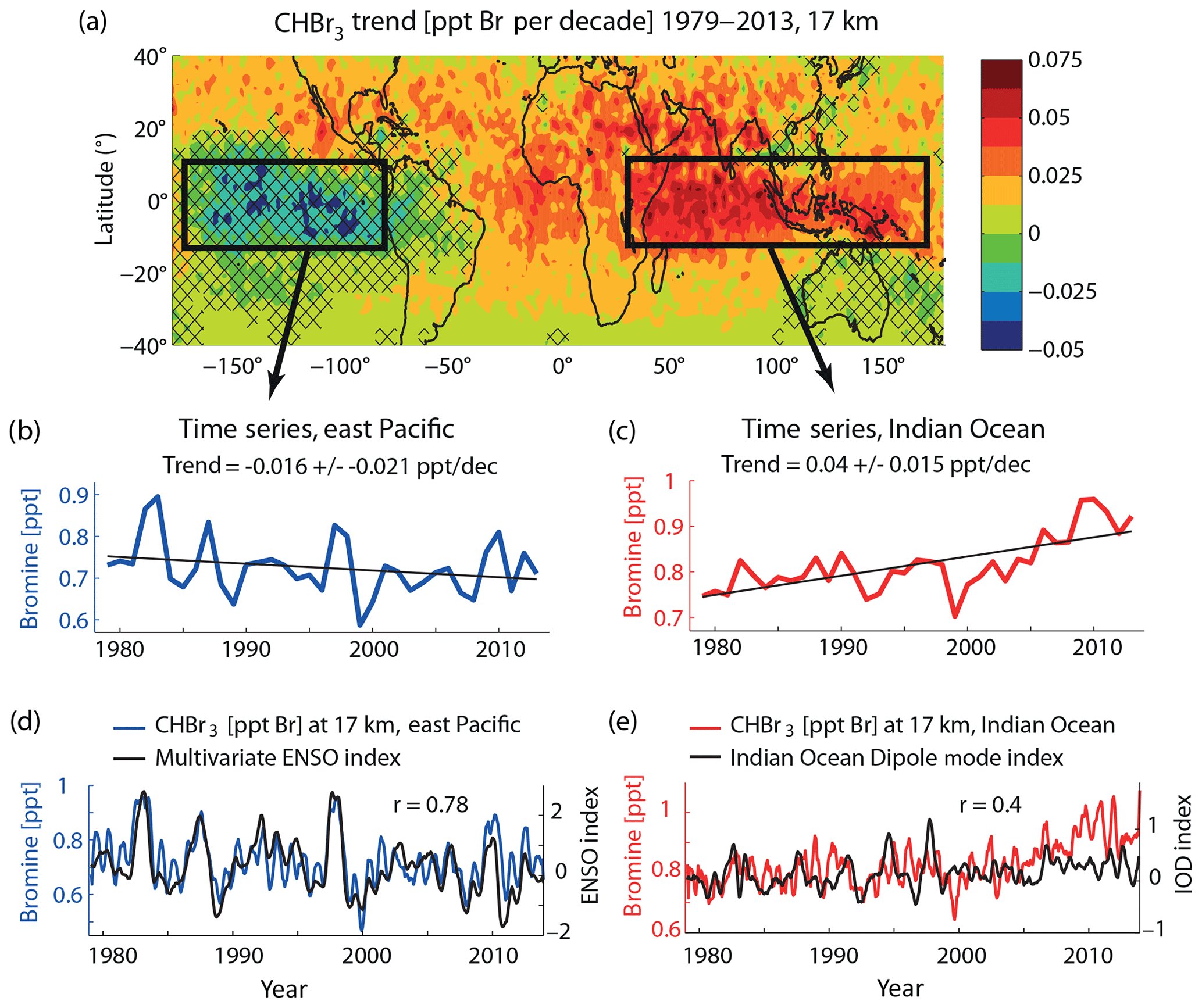
Figure 9Modelled long-term change of CHBr3 (Br per decade) at 17 km for the time period 1979–2013 (a). Time series (annual means) averaged over the east Pacific and the Indian Ocean–Maritime Continent–west Pacific region are shown together with the trend (b, c). Time series (5-month running mean) are shown together with the ENSO index and Indian Ocean Dipole index (d, e).
The projected inter-annual and long-term changes of CHBr3 injections are driven by the variability of oceanic emissions (Ziska et al., 2013), convective transport from the surface to the TTL (Aschmann et al., 2011), and transport in the TTL (Krüger et al., 2009). Our model runs are based on CHBr3 emissions that allow for changes over time due to changing meteorological surface parameters (mostly ERA-Interim) but do not take into account oceanic biogeochemical and related CHBr3 production changes. Due to increasing sea surface temperature and wind speed, CHBr3 emissions increase considerably by 7.9 % from 1979 to 2013 (Ziska et al., 2017). Changes in the modelled atmospheric transport are driven by long-term changes in ERA-Interim parameters such as temperature, winds, and humidity fields, leading to an overall trend of CHBr3 at 17 km of 10 % for 1979–2013.
The two CHBr3 time series over the east Pacific and Indian Ocean–Maritime Continent (Fig. 9b and c) show the opposite long-term behaviour but also share some of the same patterns of inter-annual variability. In particular, signals like the steep CHBr3 decrease from 1997/1998 to 1999, the increase from 2008 to 2009/2010, and the relatively high values in 1982 are common to both time series. We analyse the common and separate drivers of the variability of the two time series further by comparing them to modes of tropical climate variability.
First, we compare the time series of stratospheric bromine in the east Pacific with the Multivariate ENSO Index (MEI; Wolter and Timlin, 2011) in Fig. 9d. The irregular ENSO variations in winds and sea surface temperatures over the tropical eastern Pacific Ocean drive changes in CHBr3 emissions and atmospheric transport, leading to a high correlation of the two time series (r=0.78). During El Niño years, water in the central and eastern Pacific becomes warmer than usual and the dry and steady easterly winds turn into warm and moist westerlies, leading to an increase in the oceanic emissions. This increase is driven by meteorological and oceanic surface variations but does not allow for possible changes in biogenic CHBr3 production related to changes in the eastern Pacific upwelling system (Hepach et al., 2016). At the same time, the warm east Pacific favours stronger convection, intensifying the VSLS transport into the TTL (Aschmann et al., 2011). Overall, El Niño years lead to enhanced CHBr3 injection over the east Pacific (e.g. 1982, 1986, 1991, and 1997), while La Niña corresponds to weaker CHBr3 injection (e.g. 1988, and 2010).
Second, variations in CHBr3 at 17 km over the Indian Ocean and Maritime Continent are shown together with the Indian Ocean Dipole (IOD) mode index (Fig. 9e), an indicator of the east–west temperature gradient across the tropical Indian Ocean (Saji et al., 1999). The two time series are weakly correlated (r=0.4), sharing some of their variability. The IOD is a coupled ocean–atmosphere phenomenon with anomalous cooling of the southeastern tropical Indian Ocean and anomalous warming of the western tropical Indian Ocean during a positive phase. Associated with these changes the convection normally situated over the eastern Indian Ocean warm pool shifts to the west. For some years, the positive phase results in slightly stronger CHBr3 emissions and more effective atmospheric transport (e.g. 1982–1983, 2006). In other years, strong IOD events will not impact the CHBr3 abundance over the Indian Ocean–Maritime Continent (e.g. 1997–1998). The relatively weak correlation of CHBr3 injection and IOD results from the influence of the ENSO signal on atmospheric transport in this region. A combination of sea surface temperature (SST) anomalies in the west Indian Ocean and the ENSO signal can have varying impacts on the CHBr3 injection depending on the time of year (Fiehn et al., 2018a). While positive SST anomalies together with El Niño conditions in boreal winter and spring enhance stratospheric VSLS injection, La Niña conditions in boreal autumn can also cause stronger-than-normal stratospheric injection. Overall, the inter-annual variability of the CHBr3 time series is driven by a combination of the ocean–atmosphere modes in the Indian and Pacific Ocean; however, the strong increase during 2009–2013 is not related to either of the two modes.
The overall pattern of long-term CHBr3 changes at 17 km shows a strong similarity to the long-term changes in sea surface temperature derived from ERA-Interim data (Fig. 10). While the global mean surface temperature has increased due to anthropogenic greenhouse gas emissions (IPCC, 2007), the spatial pattern of global warming is more complex. Most regions exhibit a warming trend over the 35-year period; however, much of the eastern Pacific cooled. This cooling may either be related to an unusual strong manifestation of internal variability in the observations or be caused by external regional forcings (e.g. Wang et al., 2012; Luo et al., 2012). ERA-Interim long-term temperature changes over the oceans show good agreement with HadCRUT, a combined data set of instrumental temperature records, with only small differences (Simmons et al., 2014). Most interesting for our analysis is the correlation between the SST trends and the long-term changes of stratospheric CHBr3 entrainment. Regions with large positive SST trends such as the Indian Ocean, east Atlantic, and Maritime Continent coincide with regions where the CHBr3 entrainment trend is strongest. The east Pacific, on the other hand, stands out as the region where the SST cooling trend coincides with decreasing CHBr3 entrainment. While this relation holds for many oceanic regions, we also find outliers such as the southern Indian Ocean, where SST trends are around zero but CHBr3 entrainment shows a strong positive trend. Based on our modelling approach, the interaction of two mechanisms causes the strong correlation between the SST and CHBr3 trends. Higher sea surface temperatures and stronger surface winds force a larger flux of CHBr3 out of the ocean into the atmosphere (Ziska et al., 2013) and at the same time cause enhanced convection, transporting surface air masses into the TTL (Tegtmeier et al., 2015). As the cold point tropopause altitude shows no significant trend in radiosondes or ERA-Interim data over the 1980–2013 time period (Tegtmeier et al., 2020), CHBr3 changes at 17 km correspond directly to changes of stratospheric CHBr3 entrainment. Future SST changes can be expected to drive a continued positive trend of stratospheric CHBr3 entrainment (Hossaini et al., 2012a).
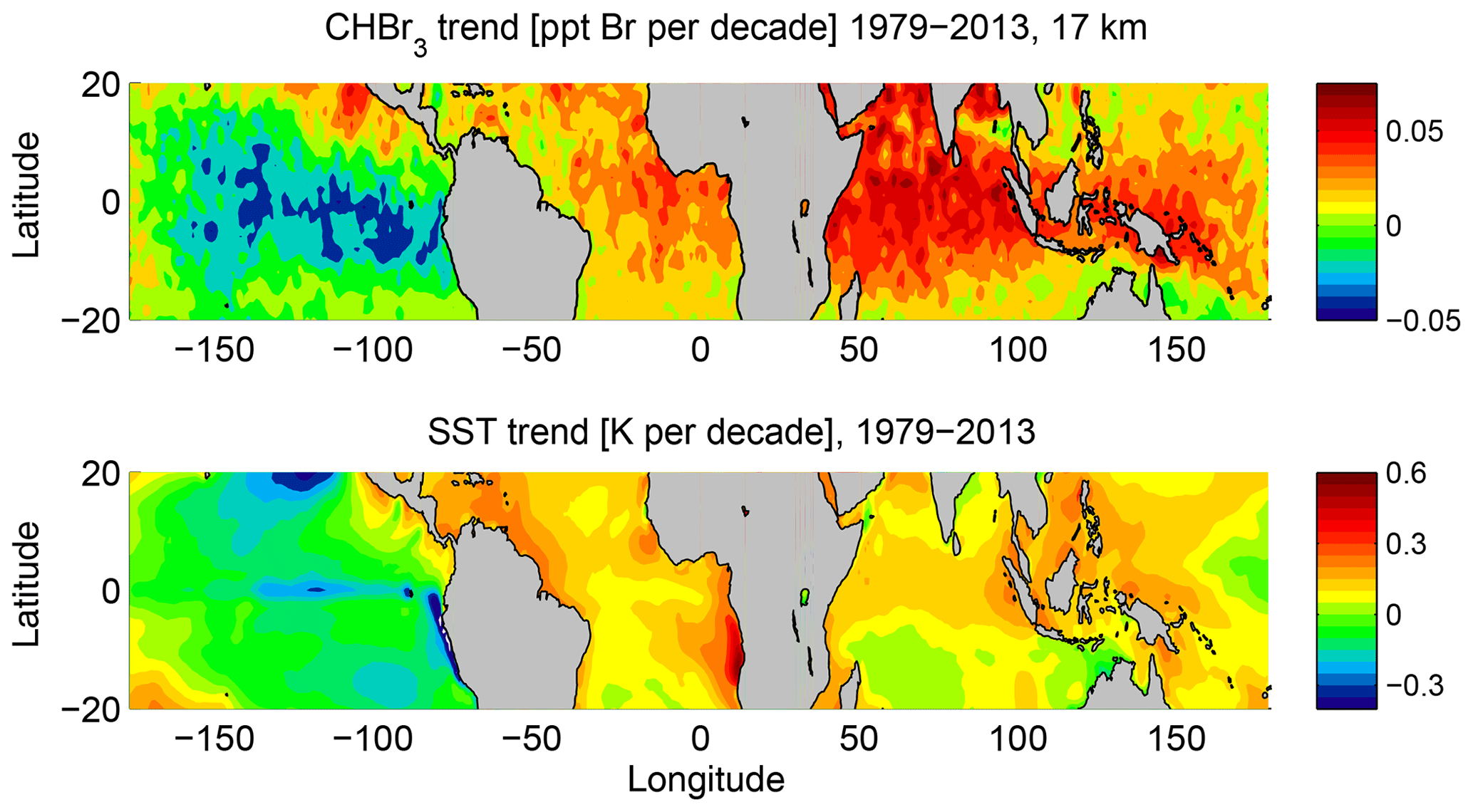
Figure 10Modelled long-term change of FLEXPART CHBr3 (ppt Br per decade) at 17 km and ERA-Interim sea surface temperature (SST) (K per decade) for the time period 1979–2013.
3.6 Overall CHBr3 and CH2Br2 contribution to stratospheric bromine
CHBr3 together with CH2Br2 provide the main contribution of oceanic bromine to the stratosphere. CH2Br2 mixing ratios in the inner tropical belt (10∘ S–10∘ N) show less variability than CHBr3, consistent with the longer lifetime, and range between 0.9 and 1.4 ppt. The largest values can be detected over the west and central Pacific and are distributed evenly over both hemispheres (Fig. 11). There is no local CH2Br2 maxima over the Indian Ocean, as observed for CHBr3, since no strong localized sources in the region exist according to the Ziska et al. (2013) climatology. However, new ship measurements in the western Indian Ocean revealed high CH2Br2 surface water concentrations, i.e. south of Madagascar in July 2011 (Fiehn et al., 2017). Seasonal and inter-annual variations in CH2Br2 are much weaker than for CHBr3, resulting in a continuous bromine entrainment into the stratosphere.
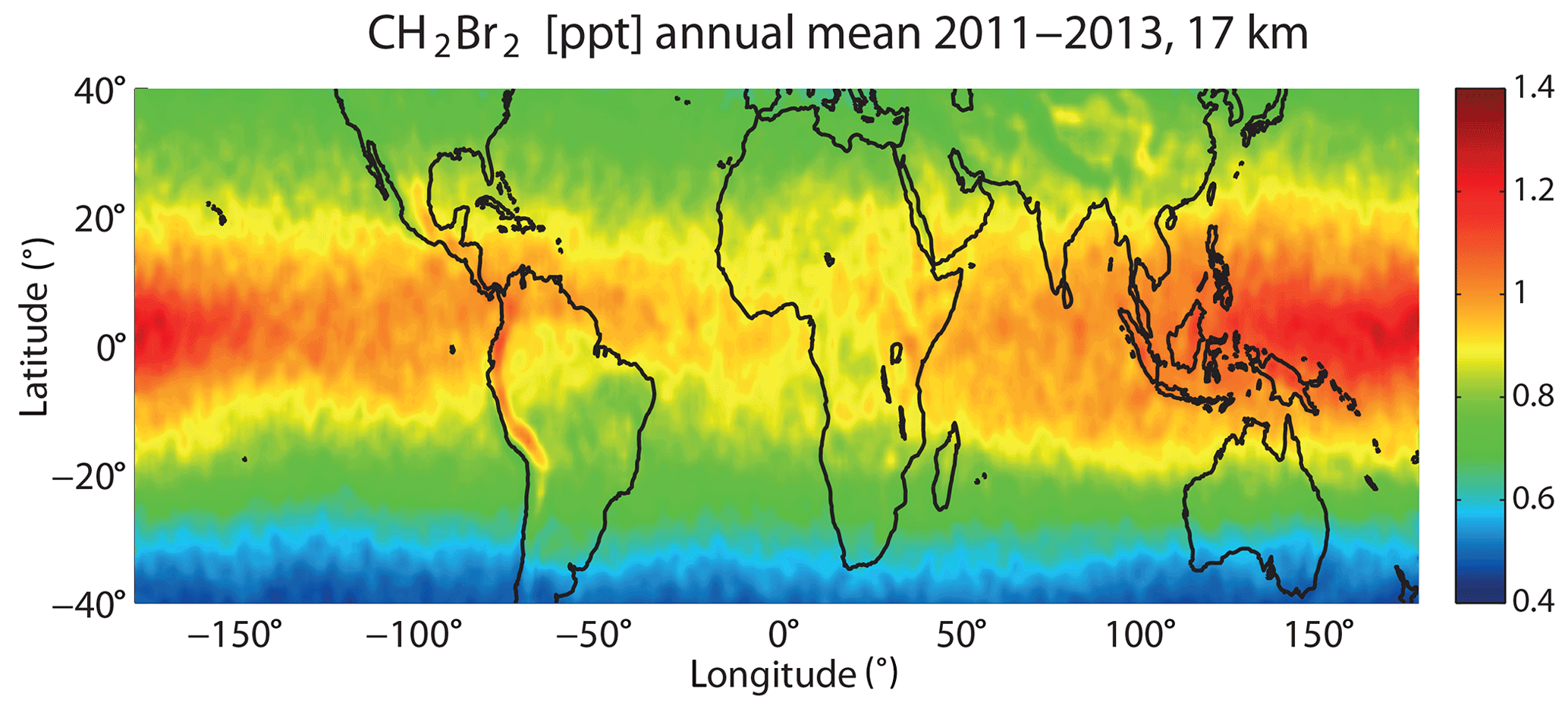
Figure 11Modelled tropical annual mean distribution of CH2Br2 (ppt) at 17 km for 2011–2013.
Figure 12 shows the annual tropical mean CHBr3 and CH2Br2 profiles averaged over 1979–2013. At the surface, tropical mean values of 1 ppt CH2Br2 and 0.6 ppt CHBr3 are simulated, which are slightly smaller than reported observations (Ziska et al., 2013, and references therein). Mixing ratios in the free troposphere decrease by nearly 50 % (10 %) for CHBr3 (CH2Br2) when compared to the marine boundary layer. Both gases are well mixed in the free troposphere with nearly constant mixing ratios of 0.3 and 0.9 ppt for CHBr3 and CH2Br2, respectively, corresponding to 0.9 and 1.8 ppt bromine (Fig. 12a). CHBr3 shows a slight S shape with elevated abundances around 12–14 km related to strong convective outflow at this level bringing marine boundary layer air directly into the lower TTL. Above 14 km, CHBr3 mixing ratios start to decrease, reaching values of 0.22 ppt at 17 km close to the cold point, corresponding to 0.66 ppt bromine. CH2Br2 mixing ratios, on the other hand, stay nearly constant up to 18 km, as expected based on its quite long lifetime of 400 to 500 d in the TTL, reaching values of 0.9 ppt (1.8 ppt bromine).
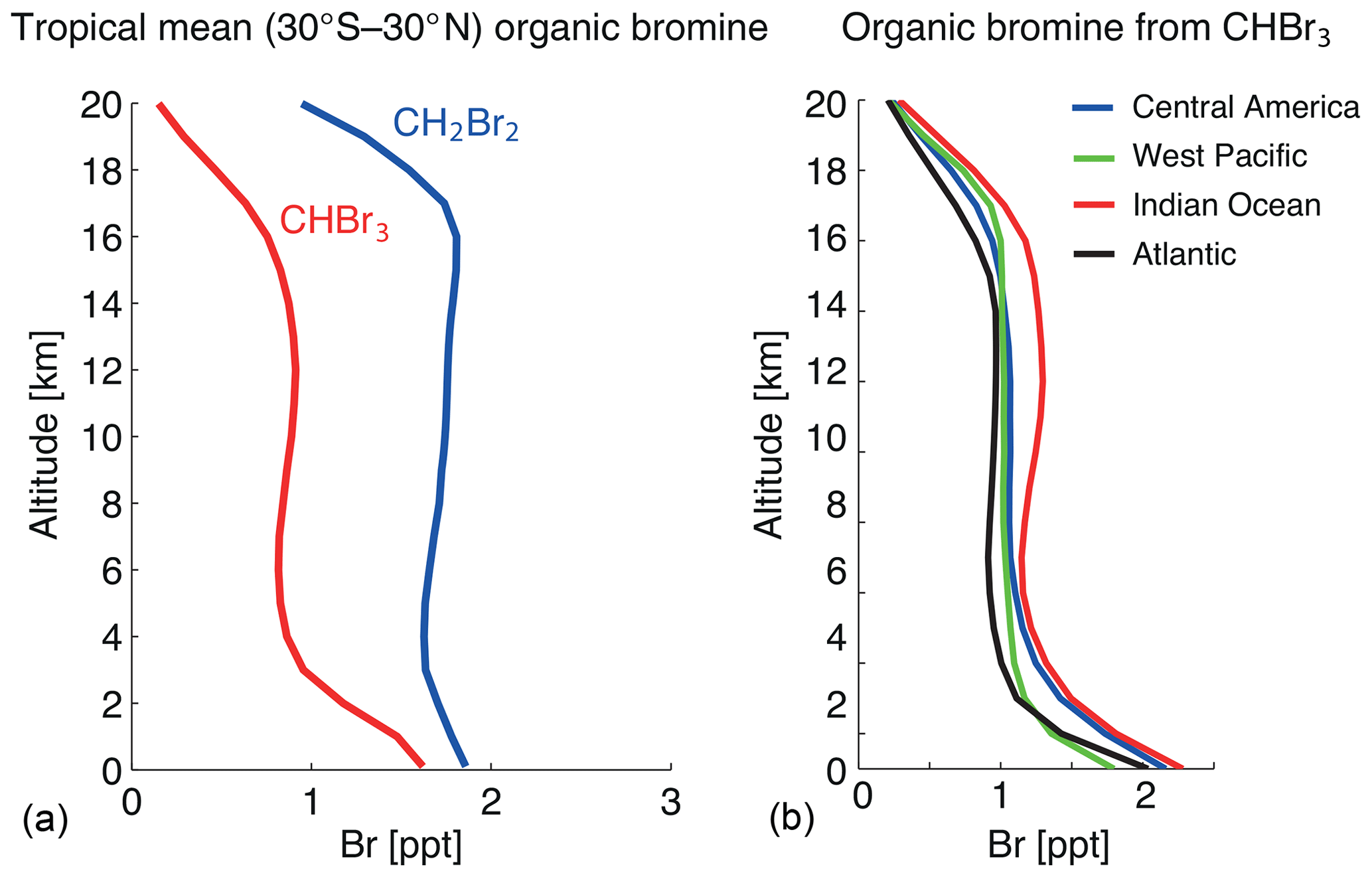
Figure 12Modelled vertical profiles of CHBr3 and CH2Br2 (ppt Br) in the tropics (30∘ S–30∘ N) (a) and of CHBr3 for Central America (0–20∘ N, 70–110∘ W), west Pacific (15∘ S–5∘ N, 140∘ E–150∘ W), Indian Ocean (0–20∘ N, 40–110∘ E), and Atlantic (0–20∘ N, 20–50∘ W) (b) for 1979–2013.
CHBr3 profiles for four different regions (Fig. 12b) show that surface atmospheric mixing ratios are strongest in the Indian Ocean and Central America. Overall, maximum mixing ratios over the Indian Ocean result from strong surface emissions combined with a relatively strong transport and main convective outflow between 11 and 14 km, giving an S-shaped CHBr3 profile. Only for the west Pacific is transport into the stratosphere more efficient; however, smaller emissions lead to the total entrainment over this region being smaller than over the Indian Ocean.
Table 2 gives the contribution of CHBr3 and CH2Br2 to the stratospheric bromine loading based on SG injection alone and based on the sum of source and PG injection. CHBr3 and CH2Br2 have been evaluated directly at the cold point (as given by ERA-Interim) and contribute 2.4 ppt Br to stratospheric bromine loading directly in the form of SG entrainment with 25 % (75 %) resulting from CHBr3 (CH2Br2). The CHBr3 estimates of 0.2 ppt (corresponding to 0.6 ppt Br) are in agreement with other studies which range from 0.1 ppt (Warwick et al., 2006; Aschmann et al., 2009) to 0.35 ppt (Hossaini et al., 2012b). For CH2Br2, our results of 0.9 ppt (corresponding to 1.8 ppt Br) agree very well with other modelling studies (Hossaini et al., 2012b) which give estimates of 0.75–0.9 ppt. The overall contribution of the two gases in the form of SG and PG entrainment of 4.7 ppt is also in good agreement with earlier studies giving estimates ranging from 4–5 ppt (Hossaini et al., 2013) to 7.7 ppt (Liang et al., 2014).
Table 2Modelled contribution of CHBr3 and CH2Br2 to the stratospheric halogen loading in the form of source gas (SG) and total (SG + PG) contribution for 2011–2013.
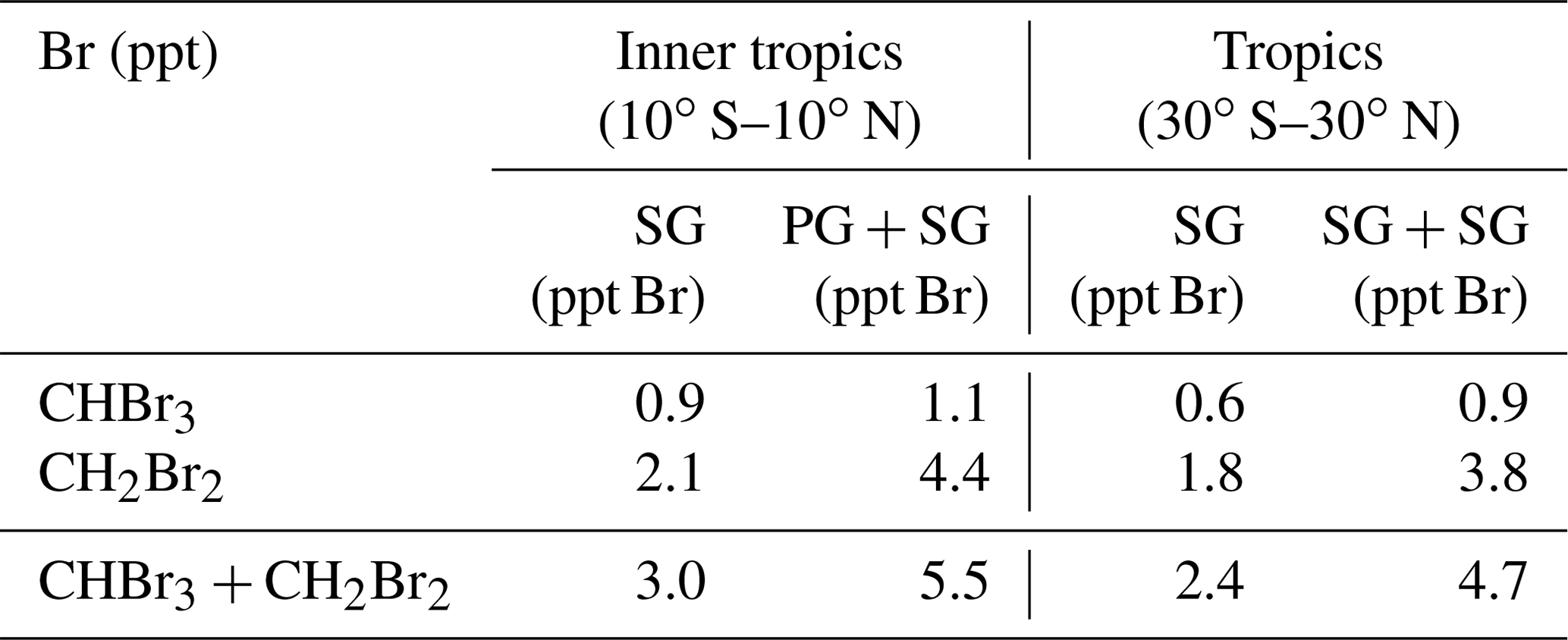
Table 3Comparison of VSLS source gas (SG) contribution derived from this study and from aircraft measurements as well as product gas (PG) contribution derived from this study and studies linking aircraft measurements and modelling.
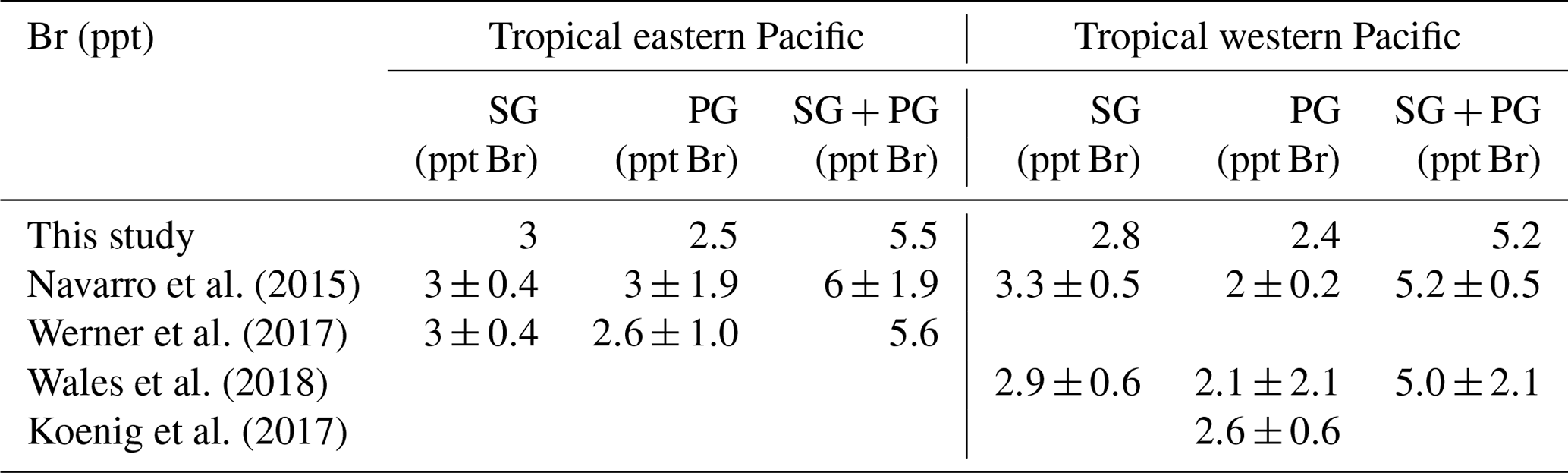
A detailed comparison of our results over the eastern and western tropical Pacific to results derived from ATTREX and CONTRAST aircraft measurements and related model calculations is given in Table 3. Considering that CHBr3 and CH2Br2 contribute >80 % of the total SG Br in the TTL, our SG estimates agree very well with the measurements (Navarro et al., 2015; Werner et al., 2017; Wales et al., 2018). PG estimates are in general characterized by larger uncertainties. The PG contribution can be inferred from atmospheric measurements of BrO, the most abundant Bry species, and the partitioning of inorganic Bry derived from a photochemical model (Koenig et al., 2017; Werner et al., 2017; Wales et al., 2018). Uncertainties in this method arise from modelling the Bry partitioning and from uncertainties in measuring BrO and can be as large as ±2.1 ppt (e.g. Wales et al., 2018).
Our study uses a simplified approach with a prescribed Bry partitioning including its spatial and temporal variations. We have carried out sensitivity studies to analyse how variations in the Bry partitioning impact the total amount of PG reaching the cold point tropopause (not shown here). Our studies show that uncertainties of 20 % in the partitioning will lead to variations of ±0.4 ppt in the PG entrainment. Such uncertainties in the Bry partitioning can result from errors in the aerosol loading and in the heterogeneous reactions. Distributions of total Bry and BrO in p-TOMCAT, the model used to derive the partitioning, have been shown to agree well with in situ and satellite observations (Yang et al., 2005, 2010). If the uncertainties in the partitioning would be as large as 50 %, the PG entrainment would show variations of ±1.1 ppt. Overall the PG entrainment based on our simplified approach agrees very well (within ±25 %) with estimates from other studies derived from BrO measurements and photochemical modelling (Table 3).
We combine observational data sets, including surface and upper-air measurements, with high-resolution atmospheric modelling in order to analyse the spatial and temporal variability of VSLS entrainment into the stratosphere. Oceanic CHBr3 in the TTL, on its way from the marine boundary layer into the stratosphere, shows a very high spatial and temporal variability. Regional maxima with mixing ratios of up to 0.4 to 0.5 ppt are simulated to be over Central America (1) and the Maritime Continent and tropical west Pacific (2), both of which are confirmed by high-altitude aircraft campaigns. The strongest stratospheric CHBr3 entrainment is projected to occur over the region of India, Bay of Bengal, and Arabian Sea (3); however, no data from aircraft campaigns are available to confirm this finding. Other tropical regions with only little convective uplift show smaller mixing ratios, mostly between 0.1 and 0.2 ppt. CHBr3 fields on daily mean or shorter timescales are characterized by pronounced spatial variations with highly localized injections.
The modelled CHBr3 maximum over Central America is caused by the co-occurrence of convectively driven short transport timescales and strong regional sources, with the latter being confirmed by data from various ship campaigns. Moreover, the combined seasonality of transport efficiency and emission strength causes the strong seasonality of CHBr3 at 17 km over Central America. The model simulations also show a high spatial variability of CHBr3 with strong latitudinal gradients, which is confirmed by available aircraft campaigns. The comparisons reveal that our model results are similar to the measurements for NH winter, but over- and underestimate (depending on the campaign) observations during NH summer, when the variability is largest. Exceptionally high CHBr3 observed during the ACCENT campaign is also evident in the model results, but only in the daily and not in the monthly mean values. Given that individual campaigns may not be representative of mean values but may rather describe one side of the large spectrum, differences between model simulations and measurements, such as the ones discussed above, have to be interpreted with caution.
The modelled CHBr3 maximum in the TTL over the west Pacific is centred south of the Equator. This distribution cannot be explained by transport timescales, which are similar north and south of the Equator and do not reveal strong hemispheric differences. Instead, strong oceanic sources south of Equator, prescribed based on limited available measurements, are responsible for the high CHBr3 mixing ratios in the SH. Measurements in the upper TTL from the ATTREX aircraft campaign show an overall good agreement with model results, but they also indicate that the model underestimates CHBr3 in the tropics. Furthermore, ATTREX measurements did not show any significant gradient between the NH and SH tropics near the tropopause. Given the scarcity of in situ measurements in the open-ocean water of the west Pacific, it may be possible that oceanic emission estimates used here are too low, especially north of the Equator. Future ship campaigns are needed to confirm spatial and temporal differences and to improve existing bottom-up emission climatologies.
The overall strongest maximum over India, Bay of Bengal, and Arabian Sea is caused by very large local sources. Transport from the ocean surface to 17 km is also efficient but not strong enough to solely explain the pronounced maxima. No upper-air measurements are available to back up this upper TTL maximum, and oceanic measurements used for the emission scenarios are also scarce. For the global tropical–extratropical distribution of CHBr3 entrainment, the largest uncertainties exist for estimated maxima in the region over India, Bay of Bengal, and Arabian Sea. In situ measurements of the oceanic sources and the atmospheric distribution are needed to reduce local uncertainties and confirm global mean values.
Our understanding of stratospheric VSLS entrainment is also limited by the fact that currently available emission inventories do not take seasonal variations in oceanic concentrations into account.
Inter-annual variability of stratospheric CHBr3 entrainment is to a large part driven by the variability of the coupled ocean–atmosphere circulation systems such as ENSO in the Pacific and IOD in the Indian Ocean. Long-term trends of the CHBr3 entrainment, on the other hand, show a pronounced correlation with the SST trends. Both relations are based on the fact that stratospheric CHBr3 entrainment is driven by strong sources and convective entrainment, which maximize for high surface temperatures and strong winds. Following the SST trends, long-term changes of CHBr3 entrainment are positive in the west Pacific and Asian monsoon region but negative in the east Pacific. The tropical mean trend accounts for an increase of 0.017±0.012 ppt Br per decade resulting in a 10 % increase over the 1979–2013 time period. The overall contribution of CHBr3 and CH2Br2 to the stratospheric halogen loading is 4.7 ppt Br with 50 % being entrained in the form of source gases and the other 50 % being entrained in the form of product gases.
The bromoform and dibromomethane emission inventory data (Ziska et al., 2013) and the FLEXPART model output can be inquired about by contacting the authors.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-7103-2020-supplement.
ST, KK, and BQ developed the idea for this paper and the model experiments. ST carried out the FLEXPART model calculations and the comparison to the aircraft observations. EA provided aircraft data. FZ compiled the Ziska et al. (2013) climatology for this study. ST wrote the manuscript with contributions from all co-authors. KK and BQ led the ROMIC THREAT project.
The authors declare that they have no conflict of interest.
Susann Tegtmeier was funded by ROMIC THREAT (01LG1217A) when compiling the study and by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – TE 1134/1 when writing the manuscript. Elliot Atlas was supported by grants from the NASA Upper Atmosphere. The authors are grateful to the ECMWF for making the reanalysis product ERA-Interim available. The authors would like to thank the editor, Jianzhong Ma, and the two anonymous reviewers for their helpful comments.
This research has been supported by the BMBF (ROMIC THREAT (grant no. 01LG1217A)).
The article processing charges for this open-access
publication were covered by a Research
Centre of the Helmholtz Association.
This paper was edited by Jianzhong Ma and reviewed by two anonymous referees.
Abbatt, J. P. D.: Interactions of Atmospheric Trace Gases with Ice Surfaces: Adsorption and Reaction, Chem. Rev., 103, 4783–4800, 2003.
Aschmann, J., Sinnhuber, B.-M., Atlas, E. L., and Schauffler, S. M.: Modeling the transport of very short-lived substances into the tropical upper troposphere and lower stratosphere, Atmos. Chem. Phys., 9, 9237–9247, https://doi.org/10.5194/acp-9-9237-2009, 2009.
Aschmann, J., Sinnhuber, B.-M., Chipperfield, M. P., and Hossaini, R.: Impact of deep convection and dehydration on bromine loading in the upper troposphere and lower stratosphere, Atmos. Chem. Phys., 11, 2671–2687, https://doi.org/10.5194/acp-11-2671-2011, 2011.
Austin, J. N. and Butchart, N.: Coupled chemistry-climate model simulations for the period 1980 to 2020: ozone depletion and the start of ozone recovery, Q. J. Roy. Meteor. Soc., 129, 3225–3249, 2003.
Braesicke, P., Keeble, J., Yang, X., Stiller, G., Kellmann, S., Abraham, N. L., Archibald, A., Telford, P., and Pyle, J. A.: Circulation anomalies in the Southern Hemisphere and ozone changes, Atmos. Chem. Phys., 13, 10677–10688, https://doi.org/10.5194/acp-13-10677-2013, 2013.
Brinckmann, S., Engel, A., Bönisch, H., Quack, B., and Atlas, E.: Short-lived brominated hydrocarbons – observations in the source regions and the tropical tropopause layer, Atmos. Chem. Phys., 12, 1213–1228, https://doi.org/10.5194/acp-12-1213-2012, 2012.
Butler, J. H., King, D. B., Lobert, J. M., Montzka, S. A., Yvon-Lewis, S. A., Hall, B. D., Warwick, N. J., Mondeel, D. J., Aydin, M., and Elkins, J. W.: Oceanic distributions and emissions of short-lived halocarbons, Global Biogeochem. Cy., 21, GB1023, https://doi.org/10.1029/2006GB002732, 2007.
Carpenter, L. J., Reimann, S., Burkholder, J. B., Clerbaux, C., Hall, B. D., Hossaini, R., Laube, J. C., and Yvon-Lewis, S. A.: Ozone-Depleting Substances (ODSs) and Other Gases of Interest to the Montreal Protocol, Chapter 1 in Scientific Assessment of Ozone Depletion: 2014, Global Ozone Research and Monitoring Project – Report No. 55, World Meteorological Organization, Geneva, Switzerland, 2014.
Chipperfield, M. P.: New version of the TOMCAT/SLIMCAT off-line chemical transport model: Intercomparison of stratospheric tracer experiments, Q. J. Roy. Meteor. Soc., 132, 1179–1203, https://doi.org/10.1256/qj.05.51, 2006.
Dee, D. P., Uppala, S. M., Simmons, A. J., Berrisford, P., Poli, P., Kobayashi, S., Andrae, U., Balmaseda, M. A., Balsamo, G., Bauer, P., Bechtold, P., Beljaars, A. C. M., van de Berg, L., Bidlot, J., Bormann, N., Delsol, C., Dragani, R., Fuentes, M., Geer, A. J., Haimberger, L., Healy, S. B., Hersbach, H., Hólm, E. V., Isaksen, L., Kållberg, P., Köhler, M., Matricardi, M., McNally, A. P., Monge-Sanz, B. M., Morcrette, J.-J., Park, B.-K., Peubey, C., de Rosnay, P., Tavolato, C., Thépaut, J.-N., and Vitart, F.: The ERA-Interim reanalysis: configuration and performance of the data assimilation system, Q. J. Roy. Meteor. Soc., 137, 553–597, https://doi.org/10.1002/qj.828, 2011.
Dorf, M., Butz, A., Camy-Peyret, C., Chipperfield, M. P., Kritten, L., and Pfeilsticker, K.: Bromine in the tropical troposphere and stratosphere as derived from balloon-borne BrO observations, Atmos. Chem. Phys., 8, 7265–7271, https://doi.org/10.5194/acp-8-7265-2008, 2008.
Engel, A., Rigby, M. A., Burkholder, J. B., Fernandez, R. P., Froidevaux, L., Hall, B. D., Hossaini, R., Saito, T., Vollmer, M. K., and Yao, B.: Update on Ozone-Depleting Substances (ODSs) and Other Gases of Interest to the Montreal Protocol, Chapter 1, in Scientific Assessment of Ozone Depletion: 2018, Global Ozone Research and Monitoring Project – Report No. 58, World Meteorological Organization, Geneva, Switzerland, 2018.
Fernandez, R. P., Salawitch, R. J., Kinnison, D. E., Lamarque, J.-F., and Saiz-Lopez, A.: Bromine partitioning in the tropical tropopause layer: implications for stratospheric injection, Atmos. Chem. Phys., 14, 13391–13410, https://doi.org/10.5194/acp-14-13391-2014, 2014.
Fiehn, A., Quack, B., Hepach, H., Fuhlbrügge, S., Tegtmeier, S., Toohey, M., Atlas, E., and Krüger, K.: Delivery of halogenated very short-lived substances from the west Indian Ocean to the stratosphere during the Asian summer monsoon, Atmos. Chem. Phys., 17, 6723–6741, https://doi.org/10.5194/acp-17-6723-2017, 2017.
Fiehn, A., Quack, B., Marandino, C. A., and Krüger, K., Transport variability of very short lived substances from the West Indian Ocean to the stratosphere, J. Geophys. Res.-Atmos., 123, 5720–5738, https://doi.org/10.1029/2017JD027563, 2018a.
Fiehn, A., Quack, B., Stemmler, I., Ziska, F., and Krüger, K.: Importance of seasonally resolved oceanic emissions for bromoform delivery from the tropical Indian Ocean and west Pacific to the stratosphere, Atmos. Chem. Phys., 18, 11973–11990, https://doi.org/10.5194/acp-18-11973-2018, 2018b.
Forster, C., Wandinger, U., Wotawa, G., James, P., Mattis, I., Althausen, D., Simmonds, P., O'Doherty, S., Jennings, S. G., Kleefeld, C., Schneider, J., Trickl, T., Kreipl, S., Jäger, H., and Stohl, A.: Transport of boreal forest fire emissions from Canada to Europe, J. Geophys. Res., 106, 22887, https://doi.org/10.1029/2001JD900115, 2001.
Forster, C., Stohl, A., and Seibert, P.: Parameterization of Convective Transport in a Lagrangian Particle Dispersion Model and Its Evaluation, J. Appl. Meteorol. Climatol., 46, 403–422, https://doi.org/10.1175/JAM2470.1, 2007.
Frenzel, A., Scheer, V., Sikorski, R., George, C., Behnke, W., and Zetzsch, C.: Heterogeneous Interconversion Reactions of BrNO2, ClNO2, Br2 and Cl2, J. Phys. Chem. A, 102, 1329–1337, 1998.
Fuhlbrügge, S., Quack, B., Tegtmeier, S., Atlas, E., Hepach, H., Shi, Q., Raimund, S., and Krüger, K.: The contribution of oceanic halocarbons to marine and free tropospheric air over the tropical West Pacific, Atmos. Chem. Phys., 16, 7569–7585, https://doi.org/10.5194/acp-16-7569-2016, 2016.
Hepach, H., Quack, B., Tegtmeier, S., Engel, A., Bracher, A., Fuhlbrügge, S., Galgani, L., Atlas, E. L., Lampel, J., Frieß, U., and Krüger, K.: Biogenic halocarbons from the Peruvian upwelling region as tropospheric halogen source, Atmos. Chem. Phys., 16, 12219–12237, https://doi.org/10.5194/acp-16-12219-2016, 2016.
Hossaini, R., Chipperfield, M. P., Monge-Sanz, B. M., Richards, N. A. D., Atlas, E., and Blake, D. R.: Bromoform and dibromomethane in the tropics: a 3-D model study of chemistry and transport, Atmos. Chem. Phys., 10, 719–735, https://doi.org/10.5194/acp-10-719-2010, 2010.
Hossaini, R., Chipperfield, M. P., Dhomse, S., Ordóñez, C., Saiz-Lopez, A., Abraham, N. L., Archibald, A. T., Braesicke, P., Telford, P., and Warwick, N.: Modelling future changes to the stratospheric source gas injection of biogenic bromocarbons, Geophys. Res. Lett., 39, L20813, https://doi.org/10.1029/2012GL053401, 2012a.
Hossaini, R., Chipperfield, M. P., Feng, W., Breider, T. J., Atlas, E., Montzka, S. A., Miller, B. R., Moore, F., and Elkins, J.: The contribution of natural and anthropogenic very short-lived species to stratospheric bromine, Atmos. Chem. Phys., 12, 371–380, https://doi.org/10.5194/acp-12-371-2012, 2012b.
Hossaini, R., Mantle, H., Chipperfield, M. P., Montzka, S. A., Hamer, P., Ziska, F., Quack, B., Krüger, K., Tegtmeier, S., Atlas, E., Sala, S., Engel, A., Bönisch, H., Keber, T., Oram, D., Mills, G., Ordóñez, C., Saiz-Lopez, A., Warwick, N., Liang, Q., Feng, W., Moore, F., Miller, B. R., Marécal, V., Richards, N. A. D., Dorf, M., and Pfeilsticker, K.: Evaluating global emission inventories of biogenic bromocarbons, Atmos. Chem. Phys., 13, 11819–11838, https://doi.org/10.5194/acp-13-11819-2013, 2013.
Hossaini, R., Chipperfield, M. P., Montzka, S. A., Rap, A., Dhomse, S., and Feng, W.: Efficiency of short-lived halogens at influencingclimate through depletion of stratospheric ozone, Nat. Geosci., 8, 186–190, https://doi.org/10.1038/ngeo2363, 2015.
Hossaini, R., Patra, P. K., Leeson, A. A., Krysztofiak, G., Abraham, N. L., Andrews, S. J., Archibald, A. T., Aschmann, J., Atlas, E. L., Belikov, D. A., Bönisch, H., Carpenter, L. J., Dhomse, S., Dorf, M., Engel, A., Feng, W., Fuhlbrügge, S., Griffiths, P. T., Harris, N. R. P., Hommel, R., Keber, T., Krüger, K., Lennartz, S. T., Maksyutov, S., Mantle, H., Mills, G. P., Miller, B., Montzka, S. A., Moore, F., Navarro, M. A., Oram, D. E., Pfeilsticker, K., Pyle, J. A., Quack, B., Robinson, A. D., Saikawa, E., Saiz-Lopez, A., Sala, S., Sinnhuber, B.-M., Taguchi, S., Tegtmeier, S., Lidster, R. T., Wilson, C., and Ziska, F.: A multi-model intercomparison of halogenated very short-lived substances (TransCom-VSLS): linking oceanic emissions and tropospheric transport for a reconciled estimate of the stratospheric source gas injection of bromine, Atmos. Chem. Phys., 16, 9163–9187, https://doi.org/10.5194/acp-16-9163-2016, 2016.
IPCC: Understanding and Attributing Climate Change, in: Climate Change 2007: The Physical Science Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Solomon, S., Qin, D., Manning, M., Chen, Z., Marquis, M., Averyt, K. B., Tignor, M., and Miller, H. L., Cambridge University Press, Cambridge, UK and New York, NY, USA, 2007.
Ko, M. K. W., Poulet, G., Blake, D. R., Boucher, O., Burkholder, J. H., Chin, M., Cox, R. A., George, C., Graf, H.-F., Holton, J. R., Jacob, D. J., Law, K. S., Lawrence, M. G., Midgley, P. M., Seakins, P. W., Shallcross, D. E., Strahan, S. E., Wuebbles, D. J., and Yokouchi, Y.: Very short-lived halogen and sulfur substances. Chapter 2 in Scientific Assessment of Ozone Depletion: 2002 Global Ozone Research and Monitoring Project – Report No. 47, World Meteorological Organization, Geneva, Switzerland, 2003.
Koenig, T. K., Volkamer, R., Baidar, S., Dix, B., Wang, S., Anderson, D. C., Salawitch, R. J., Wales, P. A., Cuevas, C. A., Fernandez, R. P., Saiz-Lopez, A., Evans, M. J., Sherwen, T., Jacob, D. J., Schmidt, J., Kinnison, D., Lamarque, J.-F., Apel, E. C., Bresch, J. C., Campos, T., Flocke, F. M., Hall, S. R., Honomichl, S. B., Hornbrook, R., Jensen, J. B., Lueb, R., Montzka, D. D., Pan, L. L., Reeves, J. M., Schauffler, S. M., Ullmann, K., Weinheimer, A. J., Atlas, E. L., Donets, V., Navarro, M. A., Riemer, D., Blake, N. J., Chen, D., Huey, L. G., Tanner, D. J., Hanisco, T. F., and Wolfe, G. M.: BrO and inferred Bry profiles over the western Pacific: relevance of inorganic bromine sources and a Bry minimum in the aged tropical tropopause layer, Atmos. Chem. Phys., 17, 15245–15270, https://doi.org/10.5194/acp-17-15245-2017, 2017.
Krüger, K. and Quack, B.: Introduction to special issue: the TransBrom Sonne expedition in the tropical West Pacific, Atmos. Chem. Phys., 13, 9439–9446, https://doi.org/10.5194/acp-13-9439-2013, 2013.
Krüger, K., Tegtmeier, S., and Rex, M.: Variability of residence time in the Tropical Tropopause Layer during Northern Hemisphere winter, Atmos. Chem. Phys., 9, 6717–6725, https://doi.org/10.5194/acp-9-6717-2009, 2009.
Liang, Q., Stolarski, R. S., Kawa, S. R., Nielsen, J. E., Douglass, A. R., Rodriguez, J. M., Blake, D. R., Atlas, E. L., and Ott, L. E.: Finding the missing stratospheric Bry: a global modeling study of CHBr3 and CH2Br2, Atmos. Chem. Phys., 10, 2269–2286, https://doi.org/10.5194/acp-10-2269-2010, 2010.
Liang, Q., Atlas, E., Blake, D., Dorf, M., Pfeilsticker, K., and Schauffler, S.: Convective transport of very short lived bromocarbons to the stratosphere, Atmos. Chem. Phys., 14, 5781–5792, https://doi.org/10.5194/acp-14-5781-2014, 2014.
Luo, J. J., Sasakia, W., and Masumoto, Y.: Indian Ocean warming modulates Pacific climate change, P. Natl. Acad. Sci. USA, 109, 18701–18706, https://doi.org/10.1073/pnas.1210239109, 2012.
Marandino, C. A., Tegtmeier, S., Krüger, K., Zindler, C., Atlas, E. L., Moore, F., and Bange, H. W.: Dimethylsulphide (DMS) emissions from the western Pacific Ocean: a potential marine source for stratospheric sulphur?, Atmos. Chem. Phys., 13, 8427–8437, https://doi.org/10.5194/acp-13-8427-2013, 2013.
McLinden, C. A., Haley, C. S., Lloyd, N. D., Hendrick, F., Rozanov, A., Sinnhuber, B.-M., Goutail, F., Degenstein, D. A., Llewellyn, E. J., Sioris, C. E., Van Rozendael, M., Pommereau, J. P., Lotz, W., and Burrows, J. P.: Odin/OSIRIS observations of stratospheric BrO: Retrieval methodology, climatology, and inferred Bry, J. Geophys. Res.-Atmos., 115, D15308, https://doi.org/10.1029/2009JD012488, 2010.
Navarro, M. A., Atlas, E. L., Saiz-Lopez, A., Rodriguez-Lloveras, X., Kinnison, D. E., Lamarque, J.-F., Tilmes, S., Filus, M., Harris, N. R., and Meneguz, E.: Airborne measurements of organic bromine compounds in the Pacific tropical tropopause layer, P. Natl. Acad. Sci. USA, 112, 13789–13793, 2015.
Nightingale, P. D., Malin, G., Law, C. S., Watson, A. J., Liss, P. S., Liddicoat, M. I., Boutin, J., and Upstill-Goddard, R. C.: In situ evaluation of air-sea gas exchange parameterizations using novel conservative and volatile tracers, Global Biogeochem. Cy., 14, 373–387, https://doi.org/10.1029/1999GB900091, 2000.
Ordóñez, C., Lamarque, J.-F., Tilmes, S., Kinnison, D. E., Atlas, E. L., Blake, D. R., Sousa Santos, G., Brasseur, G., and Saiz-Lopez, A.: Bromine and iodine chemistry in a global chemistry-climate model: description and evaluation of very short-lived oceanic sources, Atmos. Chem. Phys., 12, 1423–1447, https://doi.org/10.5194/acp-12-1423-2012, 2012.
Pisso, I., Haynes, P. H., and Law, K. S.: Emission location dependent ozone depletion potentials for very short-lived halogenated species, Atmos. Chem. Phys., 10, 12025–12036, https://doi.org/10.5194/acp-10-12025-2010, 2010.
Pyle, J. A., Warwick, N., Yang, X., Young, P. J., and Zeng, G.: Climate/chemistry feedbacks and biogenic emissions, Philos. T. R. Soc. A, 365, 1727–1740, https://doi.org/10.1098/rsta.2007.2041, 2007.
Quack, B. and Wallace, D. W. R.: Air-sea flux of bromoform: Controls, rates, and implications, Global Biogeochem. Cy., 17, GB1023, https://doi.org/10.1029/2002GB001890, 2003.
Quack, B., Atlas, E., Petrick, G., and Wallace, D. W. R.: Bromoform and dibromomethane above the Mauritanian upwelling: Atmospheric distributions and oceanic emissions, J. Geophys. Res., 112, D09312, https://doi.org/10.1029/2006JD007614, 2007.
Randel, W. J., Park, M., Emmons, L., Kinnison, D., Bernath, P., Walker, K. A., Boone, C., and Pumphrey, H.: Asian Monsoon Transport of Pollution to the Stratosphere, Science, 328, 611–613, https://doi.org/10.1126/science.1182274, 2010.
Saji, N. H., Goswami, B. N., Vinayachandran, P. N., and Yamagata, T.: A dipole made in the Tropical Indian Ocean, Nature, 401, 360–363, https://doi.org/10.1038/43854, 1999.
Salawitch, R. J.: Atmospheric chemistry: biogenic bromine, Nature, 439, 275–277, 2006.
Salawitch, R. J., Fahey, D. W., Hegglin, M. I., McBride, L. A., Tribett, W. R., and Doherty, S. J.: Twenty Questions and Answers About the Ozone Layer: 2018 Update, Scientific Assessment of Ozone Depletion: 2018, 84 pp., World Meteorological Organization, Geneva, Switzerland, 2019.
Schmidt, J. A., Jacob, D. J., Horowitz, H. M., Hu, L., Sherwen, T., Evans, M. J., Liang, Q., Sulieman, R. M., Oram, D. E., Le Breton, M., Percival, C. J., Wang, S., Dix, B., and Volkamer, R.: Modeling the observed tropospheric BrO background: Importance of multiphase chemistry and implications for ozone, OH, and mercury, J. Geophys. Res.-Atmos., 121, 11819–11835, https://doi.org/10.1002/2015JD024229, 2016.
Simmons, A. J., Poli, P., Dee, D. P., Berrisford, P., Hersbach, H., Kobayashi, S., and Peubey, C.: Estimating low-frequency variability and trends in atmospheric temperature using ERA-Interim, Q. J. Roy. Meteor. Soc., 140, 329–353, https://doi.org/10.1002/qj.2317, 2014.
Sinnhuber, B. and Meul, S.: Simulating the impact of emissions of brominated very short lived substances on past stratospheric ozone trends, Geophys. Res. Lett., 42, 2449–2456, https://doi.org/10.1002/2014GL062975, 2015.
Sioris, C. E., Kovalenko, L. J., McLinden, C. A., Salawitch, R. J., Van Roozendael, M., Goutail, F., Dorf, M., Pfeilsticker, K., Chance, K., von Savigny, C., Liu, X., Kurosu, T. P., Pommereau, J.-P., Bösch, H., and Frerick, J.: Latitudinal and vertical distribution of bromine monoxide in the lower stratosphere from Scanning Imaging Absorption Spectrometer for Atmospheric Chartography limb scattering measurements, J. Geophys. Res., 111, D14301, https://doi.org/10.1029/2005JD006479, 2006.
Stemmler, I., Hense, I., and Quack, B.: Marine sources of bromoform in the global open ocean – global patterns and emissions, Biogeosciences, 12, 1967–1981, https://doi.org/10.5194/bg-12-1967-2015, 2015.
Stohl, A. and Thomson, D. J.: A density correction for Lagrangian particle dispersion models, Bound.-Lay. Meteorol., 90, 155–167, https://doi.org/10.1023/A:1001741110696, 1999.
Stohl, A. and Trickl, T.: A textbook example of long-range transport: Simultaneous observation of ozone maxima of stratospheric and North American origin in the free troposphere over Europe, J. Geophys. Res., 104, 30445, https://doi.org/10.1029/1999JD900803, 1999.
Stohl, A., Hittenberger, M., and Wotawa, G.: Validation of the Lagrangian particle dispersion model FLEXPART against large-scale tracer experimant data, Atmos. Environ., 32, 4245–4264, 1998.
Stohl, A., Forster, C., Frank, A., Seibert, P., and Wotawa, G.: Technical note: The Lagrangian particle dispersion model FLEXPART version 6.2, Atmos. Chem. Phys., 5, 2461–2474, https://doi.org/10.5194/acp-5-2461-2005, 2005.
Stohl, A., Sodemann, H., Eckhardt, S., Frank, A., Seibert, P., and Wotawa, G.: The Lagrangian particle dispersion model FLEXPART version 8.2, Tech. rep., Norwegian Institute of Air Re- search (NILU), Kjeller, Norway, available at: http://flexpart.eu/ (last access: 2 April 2020), 2010.
Tegtmeier, S., Krüger, K., Quack, B., Atlas, E. L., Pisso, I., Stohl, A., and Yang, X.: Emission and transport of bromocarbons: from the West Pacific ocean into the stratosphere, Atmos. Chem. Phys., 12, 10633–10648, https://doi.org/10.5194/acp-12-10633-2012, 2012.
Tegtmeier, S., Krüger, K., Quack, B., Atlas, E., Blake, D. R., Boenisch, H., Engel, A., Hepach, H., Hossaini, R., Navarro, M. A., Raimund, S., Sala, S., Shi, Q., and Ziska, F.: The contribution of oceanic methyl iodide to stratospheric iodine, Atmos. Chem. Phys., 13, 11869–11886, https://doi.org/10.5194/acp-13-11869-2013, 2013.
Tegtmeier, S., Ziska, F., Pisso, I., Quack, B., Velders, G. J. M., Yang, X., and Krüger, K.: Oceanic bromoform emissions weighted by their ozone depletion potential, Atmos. Chem. Phys., 15, 13647–13663, https://doi.org/10.5194/acp-15-13647-2015, 2015.
Tegtmeier, S., Anstey, J., Davis, S., Dragani, R., Harada, Y., Ivanciu, I., Pilch Kedzierski, R., Krüger, K., Legras, B., Long, C., Wang, J. S., Wargan, K., and Wright, J. S.: Temperature and tropopause characteristics from reanalyses data in the tropical tropopause layer, Atmos. Chem. Phys., 20, 753–770, https://doi.org/10.5194/acp-20-753-2020, 2020.
Tissier, A.-S. and Legras, B.: Convective sources of trajectories traversing the tropical tropopause layer, Atmos. Chem. Phys., 16, 3383–3398, https://doi.org/10.5194/acp-16-3383-2016, 2016.
Toon, O. B., Starr, D. O., Jensen, E. J., Newman, P. A., Platnick, S., Schoeberl, M. R., Wennberg, P. O., Wofsy, S. C., Kurylo, M. J., Maring, H., Jucks, K. W., Craig, M. S., Vasques, M. F., Pfister, L., Rosenlof, K. H., Selkirk, H. B., Colarco, P. R., Kawa, S. R., Mace, G. G., Minnis, P., and Pickering, K. E.: Planning, implementation, and first results of the Tropical Composition, Cloud and Climate Coupling Experiment (TC4), J. Geophys. Res., 115, D00J04, https://doi.org/10.1029/2009JD013073, 2010.
von Glasow, R., von Kuhlmann, R., Lawrence, M. G., Platt, U., and Crutzen, P. J.: Impact of reactive bromine chemistry in the troposphere, Atmos. Chem. Phys., 4, 2481–2497, https://doi.org/10.5194/acp-4-2481-2004, 2004.
Wales, P. A., Salawitch, R. J., Nicely, J. M., Anderson, D. C., Canty, T. P., Baidar, S., Dix, B., Koenig, T. K., Volkamer, R., Chen, D., Huey, L. G., Tanner, D. J., Cuevas, C. A., Fernandez, R. P., Kinnison, D. E., Lamarque, J.-F., Saiz-Lopez, A., Atlas, E. L., Hall, S. R., Navarro, M. A., Pan, L. L., Schauffler, S. M., Stell, M., Tilmes, S., Ullmann, K., Weinheimer, A. J., Akiyoshi, H., Chipperfield, M. P., Deushi, M., Dhomse, S. S., Feng, W., Graf, P., Hossaini, R., Jöckel, P., Mancini, E., Michou, M., Morgenstern, O., Oman, L. D., Pitari, G., Plummer, D. A., Revell, L. E., Rozanov, E., Saint-Martin, D., Schofield, R., Stenke, A., Stone, K. A., Visioni, D., Yamashita, Y., and Zeng, G.: Stratospheric injection of brominated very short-lived substances: Aircraft observations in the Western Pacific and representation in global models, J. Geophys. Res.-Atmos., 123, 5690–5719, https://doi.org/10.1029/2017JD027978, 2018.
Wang, B., Liu, J., Kim, H. J., Webster, P. J., and Yim, S. Y.: Recent change of the global monsoon precipitation (1979–2008), Clim. Dynam., 39, 1123–1135, https://doi.org/10.1007/s00382-011-1266-z, 2012.
Warwick, N. J., Pyle, J. A., Carver, G. D., Yang, X., Savage, N. H., O'Connor, F. M., and Cox, R. A.: Global modeling of biogenic bromocarbons, J. Geophys. Res., 111, D24305, https://doi.org/10.1029/2006JD007264, 2006.
Werner, B., Stutz, J., Spolaor, M., Scalone, L., Raecke, R., Festa, J., Colosimo, S. F., Cheung, R., Tsai, C., Hossaini, R., Chipperfield, M. P., Taverna, G. S., Feng, W., Elkins, J. W., Fahey, D. W., Gao, R.-S., Hintsa, E. J., Thornberry, T. D., Moore, F. L., Navarro, M. A., Atlas, E., Daube, B. C., Pittman, J., Wofsy, S., and Pfeilsticker, K.: Probing the subtropical lowermost stratosphere and the tropical upper troposphere and tropopause layer for inorganic bromine, Atmos. Chem. Phys., 17, 1161–1186, https://doi.org/10.5194/acp-17-1161-2017, 2017.
Wolter, K. and Timlin, M. S.: El Niño/Southern Oscillation behaviour since 1871 as diagnosed in an extended multivariate ENSO index (MEI.ext). Int. J. Climatol., 31, 1074–1087, https://doi.org/10.1002/joc.2336, 2011.
Yamamoto, H., Yokouchi, Y., Otsuki, A., and Itoh, H.: Depth profiles of volatile halogenated hydrocarbons in seawater in the Bay of Bengal, Chemosphere, 45, 371–377, https://doi.org/10.1016/s0045-6535(00)00541-5, 2001.
Yang, X., Cox, R. A., Warwick, N. J., Pyle, J. A., Carver, G. D., O'Connor, F. M., and Savage, N. H.: Tropospheric bromine chemistry and its impacts on ozone: A model study, J. Geophys. Res., 110, D23311, https://doi.org/10.1029/2005JD006244, 2005.
Yang, X., Pyle, J. A., Cox, R. A., Theys, N., and Van Roozendael, M.: Snow-sourced bromine and its implications for polar tropospheric ozone, Atmos. Chem. Phys., 10, 7763–7773, https://doi.org/10.5194/acp-10-7763-2010, 2010.
Yang, X., Abraham, N. L., Archibald, A. T., Braesicke, P., Keeble, J., Telford, P. J., Warwick, N. J., and Pyle, J. A.: How sensitive is the recovery of stratospheric ozone to changes in concentrations of very short-lived bromocarbons?, Atmos. Chem. Phys., 14, 10431–10438, https://doi.org/10.5194/acp-14-10431-2014, 2014.
Ziska, F., Quack, B., Abrahamsson, K., Archer, S. D., Atlas, E., Bell, T., Butler, J. H., Carpenter, L. J., Jones, C. E., Harris, N. R. P., Hepach, H., Heumann, K. G., Hughes, C., Kuss, J., Krüger, K., Liss, P., Moore, R. M., Orlikowska, A., Raimund, S., Reeves, C. E., Reifenhäuser, W., Robinson, A. D., Schall, C., Tanhua, T., Tegtmeier, S., Turner, S., Wang, L., Wallace, D., Williams, J., Yamamoto, H., Yvon-Lewis, S., and Yokouchi, Y.: Global sea-to-air flux climatology for bromoform, dibromomethane and methyl iodide, Atmos. Chem. Phys., 13, 8915–8934, https://doi.org/10.5194/acp-13-8915-2013, 2013.
Ziska, F., Quack, B., Tegtmeier, S., Stemmler, I., and Krüger, K.: Future emissions of marine halogenated very-short lived substances under climate change, J. Atmos. Chem., 74, 245, https://doi.org/10.1007/s10874-016-9355-3, 2017.