the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 09 Apr 2020
| 09 Apr 2020
Oxygen and sulfur mass-independent isotopic signatures in black crusts: the complementary negative Δ33S reservoir of sulfate aerosols?
Isabelle Genot
David Au Yang
Erwan Martin
Pierre Cartigny
Erwann Legendre
Marc De Rafelis
To better understand the formation and the oxidation pathways leading to gypsum-forming “black crusts” and investigate their bearing on the whole atmospheric SO2 cycle, we measured the oxygen (δ17O, δ18O, and Δ17O) and sulfur (δ33S, δ34S, δ36S, Δ33S, and Δ36S) isotopic compositions of black crust sulfates sampled on carbonate building stones along a NW–SE cross section in the Parisian basin. The δ18O and δ34S values, ranging between 7.5 ‰ and 16.7±0.5 ‰ (n=27, 2σ) and between −2.66 ‰ and 13.99±0.20 ‰, respectively, show anthropogenic SO2 as the main sulfur source (from ∼2 % to 81 %, average ∼30 %) with host-rock sulfates making the complement. This is supported by Δ17O values (up to 2.6 ‰, on average ∼0.86 ‰), requiring > 60 % of atmospheric sulfates in black crusts. Negative Δ33S and Δ36S values between −0.34 ‰ and 0.00±0.01 ‰ and between −0.76 ‰ and ‰, respectively, were measured in black crust sulfates, which is typical of a magnetic isotope effect that would occur during the SO2 oxidation on the building stone, leading to 33S depletion in black crust sulfates and subsequent 33S enrichment in residual SO2. Except for a few samples, sulfate aerosols mostly have Δ33S values > 0 ‰, and no processes can yet explain this enrichment, resulting in an inconsistent S budget: black crust sulfates could well represent the complementary negative Δ33S reservoir of the sulfate aerosols, thus solving the atmospheric SO2 budget.
- Article
(13440 KB) -
Supplement
(588 KB) - BibTeX
- EndNote





The oxidation of sulfur dioxide emitted into the atmosphere (between 100 and 110 Tg(SO2) yr−1; Klimont et al., 2013) can result in the formation of H2SO4 that forms sulfate aerosols; having light-scattering properties that alter the radiative balance of the planet. Furthermore, they also modify the microphysical properties of clouds through the number and size of cloud condensation nuclei (CCN; e.g., Weber et al., 2001). Although quantified with large uncertainties, the formation of sulfate aerosols results in an Earth surface cooling (Forster et al., 2007), with a negative radiative forcing from −0.62 to −0.21 W m−2 (on average W m−2). Overall, sulfate aerosols are the most efficient particles that counterbalance the greenhouse effect (Stocker, 2014). Uncertainties regarding the formation of sulfate aerosols relate to the large variety of oxidants and conditions (e.g., pH), but, in view of their major impact on climate, a more accurate understanding of the formation of these particles is necessary.
Primary sulfate aerosols consist of sulfates formed during their emission into the atmosphere (e.g., sea-salt sulfates, combustion products, volcanic sulfates), which involve therefore a local origin (Holt and Kumar, 1991). Secondary sulfate aerosols are formed later in the atmosphere following various oxidation pathways (e.g., oxidation via OH, O2 transition-metal ion (TMI), O3, H2O2, and NO2) and relate to a local or more distant sulfur source (Seinfeld and Pandis, 2016). SO2 oxidation can occur in the gas phase (homogeneous reaction, for example, with OH), in the aqueous phase (e.g., with H2O2, O3, O2 TMI), or on a surface (heterogeneous reaction), resulting in different sizes and number of aerosol particles with distinct effects on radiative balance.
Stable isotope geochemistry is a central tool for both characterizing sulfur sources and quantifying the different oxidants. The δ notation used here is defined as
And isotope fractionation factors are expressed as follows:
with A and B being two different phases.
Given that the oxidants have distinct δ18O and Δ17O signatures, the SO2 oxidation pathways are commonly constrained using oxygen multi-isotope ratios (δ18O, δ17O, and Δ17O, defined in the following section) (Alexander et al., 2012; Bindeman et al., 2007; Jenkins and Bao, 2006; Lee and Thiemens, 2001; Savarino et al., 2000; Martin, 2018). Sulfur isotope fractionation during SO2 oxidation by OH, O2 TMI, H2O2, O3 (Harris et al., 2012a, b, 2013a, b), and NO2 (Au Yang et al., 2018) have been determined, so additional constraints can also be brought by S multi-isotope compositions (δ34S, δ33S, δ36S, Δ33S, and Δ36S). At present, however, it is difficult to reach a consistent budget for tropospheric SO2 oxidation (chemically and isotopically). Indeed, most of rural and urban sulfate aerosols have positive Δ33S values (Au Yang et al., 2019; Guo et al., 2010; Han et al., 2017; Lin et al., 2018b; Romero and Thiemens, 2003; Shaheen et al., 2014), implying either a source of SO2 with Δ33S > 0 ‰ (which has not been identified yet as all known sources have Δ33S ∼0 ‰; Lin et al., 2018b) or more likely processes such as SO2 photolysis in the stratosphere (e.g., Farquhar et al., 2001), contributing to the 33S-enriched tropospheric sulfate reservoir from initial SO2 with Δ33S =0 ‰, which should be balanced by a 33S-depleted reservoir but which remains scarce (see Shaheen et al., 2014; Han et al., 2017; Lin et al., 2018a). Negative Δ33S values were suggested to result specifically from combustion (Lee et al., 2002) and/or OCS (carbonyl sulfide) photolysis (Lin et al., 2011). Still, the mass balance among positive and negative Δ33S values is not consistent. As none of the most significant tropospheric SO2 oxidation reactions can account for Δ33S anomalies in sulfate aerosols (Au Yang et al., 2018; Guo et al., 2010; Han et al., 2017; Harris et al., 2013b; Lee et al., 2002; Lin et al., 2018a, b; Romero and Thiemens, 2003; Shaheen et al., 2014), this leads to the suggestion that either some reactions or SO2 sources have been overlooked. Finally, a recent study highlights the possibility of SO2 oxidation on mineral dust surfaces resulting in 33S-depleted sulfate deposition in rural environments and subsequent 33S enrichment of residual SO2 transported to cities (Au Yang et al., 2019), but the negative Δ33S values are still missing.
In this respect, black crusts potentially represent new ways to sample the atmosphere in urban regions at a relatively global scale. They are generally formed by the sulfation of the underlying carbonate substrate resulting in a gypsum layer (Camuffo, 1995) (Fig. 1). Due to their degradation effects on monuments and buildings, in particular because the molar volume of CaSO4 is larger than that of CaCO3, several studies investigated sources of sulfur in black crusts, using primarily the isotopic composition of sulfur (δ34S) and oxygen (δ18O) and microscopic and mineralogical aspects. Anthropogenic sulfur was found to be the major source contributing to monument degradation in several localities compared to marine or volcanic sulfate sources (Longinelli and Bartelloni, 1978; Montana et al., 2008, 2012; Torfs et al., 1997). Sulfates from the host rock, i.e., plaster, mortar, or oxidized pyrite (defined as intrinsic in the literature; Klemm and Siedel, 2002; Kloppmann et al., 2011; Kramar et al., 2011; Vallet et al., 2006), and sulfates from aquifer rising by capillarity (Kloppmann et al., 2014) were also identified as sulfur sources in black crusts. Black crusts being sometimes the host of microbial activity (Gaylarde et al., 2007; Sáiz-Jiménez, 1995; Scheerer et al., 2009; Schiavon, 2002; Tiano, 2002), other studies investigated the role of bacteria in gypsum formation through sulfate reduction and/or SO2 oxidation (Tiano, 2002; Tiano et al., 1975). Except the work of Šrámek (1980) measuring black crust sulfates δ34S that rule out the implication of micro-organisms in their formation, no further constraint has been brought so far. In this paper, we present new oxygen and sulfur isotopic composition measurements of sulfate extracted from black crusts and report significant Δ17O, Δ33S, and Δ36S anomalies that help to discuss oxygen and sulfur isotopic variations both in term of source effects to elucidate their origin and in terms of fractionation processes leading to black crust formation in the Paris area.

Figure 1Thin layer of black crusts formed on a carbonate building stone on a church wall in Fécamp city.
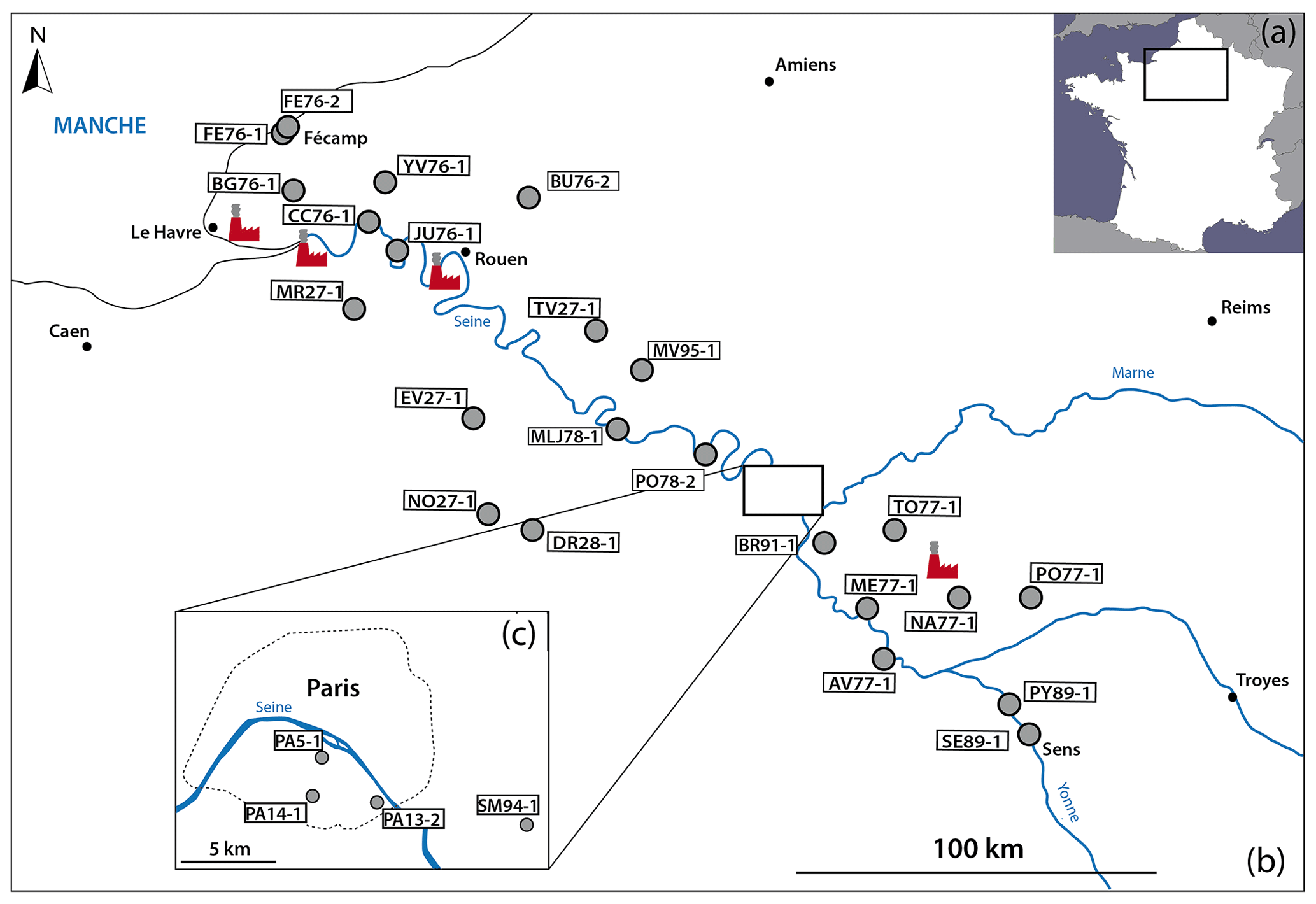
Figure 2Maps with the sampling locations. (a) Location of the studied area in the northern Paris Basin on the map of France. (b) The NW–SE cross section from Fécamp to Sens with the 27 samples and the four power plants (in red). (c) Zoomed-in view of the samples located in the Paris area.
As with many chemical reactions, O and S isotopic compositions of SO2 vary during their oxidation. Most reactions are “mass-dependent”, meaning the isotopic fractionation relies on the mass differences between the isotopes; this remains valid for most unidirectional (kinetic) and/or exchange (equilibrium) reactions. In a system with at least three isotopes, mass fractionation laws at equilibrium and high temperature can be derived from their partition function (Bigeleisen and Mayer, 1947; Dauphas and Schauble, 2016; Urey, 1947; Young et al., 2002) as follows, for instance, with oxygen isotopes and SO2 oxidation in sulfates:
with being the mass exponent describing the relative fractionation between 17O∕16O and 18O∕16O, m being the mass of each isotope, and being the isotope fractionation factor between two phases (defined in the introduction). The same equations can be written for 33S and 36S.
Here, we will use 17β, 33β, and 36β values (∼0.5305, 0.515 and 1.889, respectively) for the high-temperature limit, which has been shown to be applicable for a wide range of temperature phases (Dauphas and Schauble, 2016) and isotope systems (S, Fe, Mg, O, Si, etc.). Thus, the β exponent represents the slope in a δ-δ space, called the “mass-dependent fractionation line”, which is actually approximated from a curve (this simplification is not used in this paper). Deviations from the reference “mass-dependent” curve do not imply necessarily isotopic variations that are independent from the isotope masses. These deviations are quantified with the Δ parameter expressed by Eqs. (1), (2), and (3) (Farquhar and Wing, 2003; Thiemens, 1999):
Small nonzero Δ17O–Δ33S–Δ36S values (typically between −0.1 ‰ and +0.1 ‰) can result from mixing, mass-dependent processes such as Rayleigh distillation, or mass conservation effects and nonequilibrium processes (Farquhar et al., 2007a; Ono et al., 2006a), whereas large nonzero Δ17O−Δ33S–Δ36S values (higher than +0.2 ‰ or lower than −0.2 ‰) imply mass-independent fractionation (Cabral et al., 2013; Delavault et al., 2016; Farquhar et al., 2000, 2007b, 2002; Ono et al., 2003). Oxidation reactions would then change δ17O and δ18O but not the Δ17O, which would primarily vary through mixing of O reservoirs with variable Δ17O. Possible mechanisms producing nonzero Δ17O–Δ33S–Δ36S values recorded in sulfate aerosols are discussed in the following sections. In this paper, we investigate the different processes responsible for the Δ17O, Δ33S, and Δ36S values recorded by black crust sulfates and what can be inferred in terms of black crust formation.
3.1 Sampling sites
To access sulfate aerosols from the Parisian region, black crusts were sampled following the prevailing winds according to a NW–SE cross section from Fécamp to Sens (Fig. 2b, c). Therefore, the studied area covers rural, urban, and industrial zones including four power plants, major highways, and the large Paris metropolis.
Table 1Characteristics of black crust samples. Their name was given according to the city and the department where they are located and following the number of samples gathered at the same place (NA77-1: NA = Nangis; 77= department; -1= first sample collected).

A total of 27 samples were collected on the external faces of churches and monuments and on walls in the streets. The substrates were generally Lutetian and Cretaceous limestone, i.e., the typical building rocks in the Parisian basin. To ensure a representative sample of sulfate aerosols, the sampling was carried out preferentially oriented to NW or, if possible, not directly exposed to traffic emissions. Moreover, to avoid sulfate contamination from soils (i.e., salts by capillary action, water from runoff), black crusts were sampled at least at a height of 1.50 m above ground level. More details about the samples are summarized in Table 1.
3.2 Methods
An X-ray diffractometer (XRD D2 phaser BRUKER, ISTeP Sorbonne Université) was used to specify the mineralogical nature of each sample and therefore to demonstrate the nature of sulfur. Structural and chemical aspects were subsequently investigated using scanning electron microscopy (SEM, ISTeP Sorbonne Université).
Sulfates were leached from 20 to 100 mg of black crusts and the conversion of gypsum into pure barite was performed according to the protocol developed at the Institut des Sciences de la Terre de Paris (ISTeP) as described by Le Gendre et al. (2017). The use of an ion-exchange resin in this protocol enables the concentration and separation of sulfates from other compounds such as nitrates that can affect the O isotopic measurements. From about 3 mg of the pure barite samples, the sulfate O isotopic ratios were measured using the laser fluorination line coupled to a Delta V IR-MS (isotope ratio mass spectrometer) at the Institut de Physique du Globe de Paris (IPGP) (Bao and Thiemens, 2000). Due to SO2F2 formation during BaSO4 fluorination, which leads to incomplete O2 extraction, measured δ18O and δ17O are fractionated but were corrected as deduced from the analysis of the international barite standard NBS127 (δ18O =8.6 ‰, Δ17O ∼0 ‰); no correction was applied to Δ17O, as it remained unchanged (Bao and Thiemens, 2000). For two NBS127 measurements measured each day during 5 d (n=10), we obtained a mean δ18O (2σ) and a mean Δ17O (2σ) within error of the recent value reported by Cowie and Johnston (2016). Bao (2006) reported up to 2 ‰ variation in the correction factor that would result from sample impurity, but, as our samples were purified with an ion-exchange resin and the mean variation of the duplicates is lower than for NBS127, we applied a correction factor of 9.03 to δ18O for all analyzed samples based on the certified value of NBS127.
The remaining BaSO4 was reduced to hydrogen sulfide (H2S) by reaction for 2 h with a heated mixture of hydrochloric (HCl), hydroiodic (HI), and hypophosphorous (H3PO2) acids following the protocol described in Thode et al. (1961). H2S was purged and precipitated as silver sulfide (Ag2S) by passing through a silver nitrate (AgNO3) solution. Ag2S was then converted to SF6, purified (Ono et al., 2006b), and quantified before being analyzed by isotope ratio mass spectrometry (Thermo Fisher MAT-253) at McGill University. The δ34S values are expressed versus V-CDT (Vienna-Canyon Diablo Troilite) assuming a vs. CDT isotope composition. Our data were then expressed against CDT following the method described by Defouilloy et al. (2016). Analysis of the IAEA-S1 in the laboratory yielded the following: δ34S , Δ33S =0.09 ‰, and Δ36S ‰ vs. V-CDT. Analysis of the IAEA-S3 (n=8) gave the following: δ34S ‰, Δ33S 3 ‰, and Δ36S ‰ vs. V-CDT. All values are within the ranges of δ34S, Δ33S, and Δ36S accepted or measured by other laboratories for these international standards (Au Yang et al., 2016; Farquhar et al., 2007b; Labidi et al., 2014; Ono et al., 2006b; Geng et al., 2019).
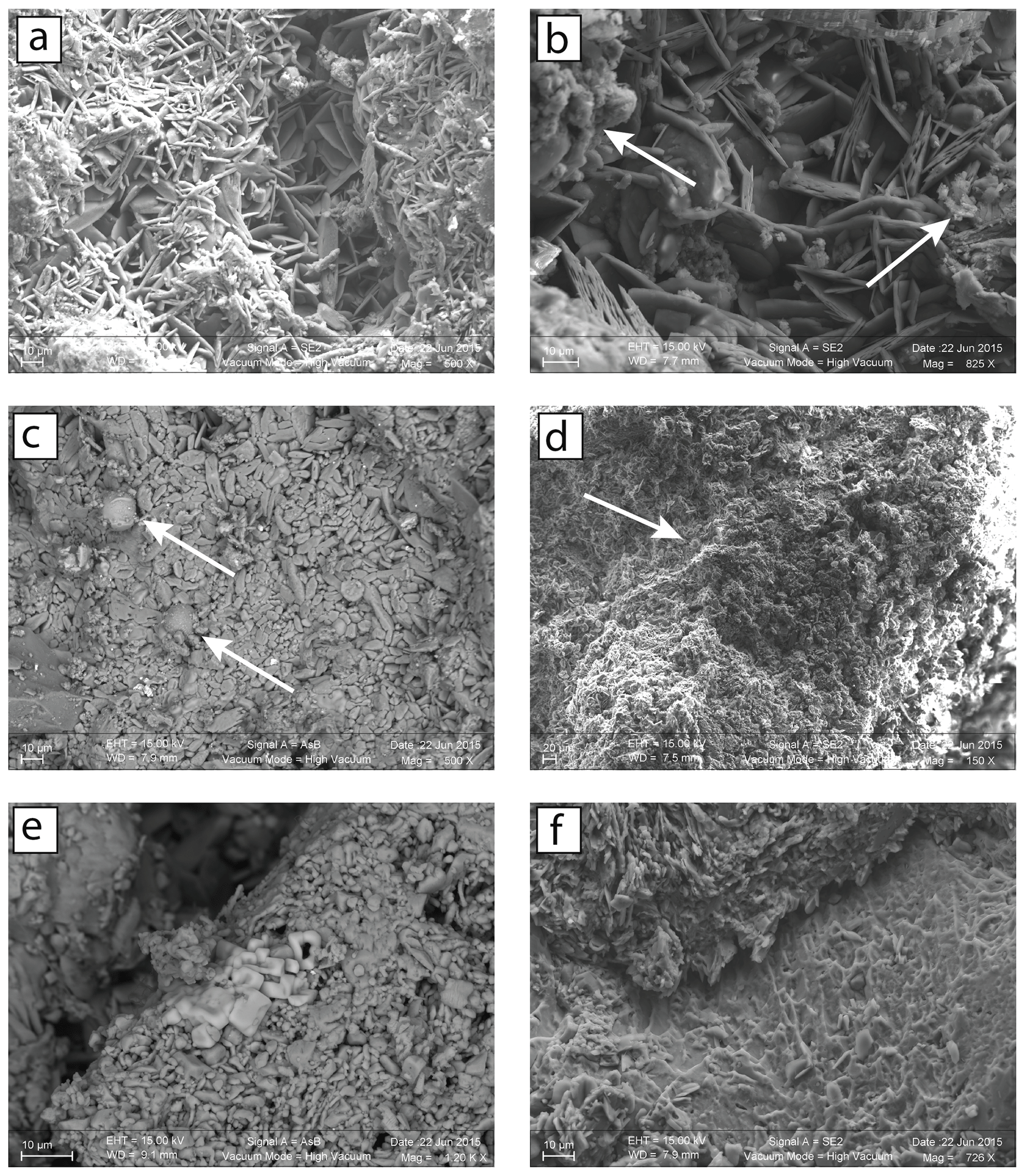
Figure 3SEM images of black crust samples from Paris (PA14-1, PA13-2) and Montfort-sur-Risles (MR27-1). (a) Two distinct layers in the crust: an upper opaque one with aggregates of particulate matter and clay minerals (left and right sides of the picture) and a more crystallized one with acicular gypsum crystals perpendicular to the host substrate. (b) There is a presence of soot (arrow) on the two layers (PA14-1). (c) Fly ashes (arrow) with the formation of small gypsum crystals are on their surfaces (PA13-2). (d) Large amount of fly ashes (arrow and smaller not indicated) and soot is in the MR27-1 sample, originating from a rural place. (e) Isolated cubic crystals of halite (NaCl) in MR27-1 sample, originating form 28 km from the coastline. (f) Dissolution of the underlying limestone (on the bottom) and subsequently the precipitation of gypsum (on the top).
4.1 Morphological and chemical aspects
After having confirmed the gypsum nature of the sample by X-ray diffraction, the structural and chemical aspects of black crusts from four different environments were investigated on the basis of SEM observations. In agreement with previous studies (Fronteau et al., 2010; Siegesmund et al., 2007), all samples display two distinct layers. An opaque layer (a few tens of micrometers thick) comprising massive and sparse gypsum crystals as well as aggregates of clay minerals and particulate matter was overlying a layer (∼100 µm) composed of more crystallized acicular and rosette-like crystal gypsum (tens of micrometers, Fig. 3a). As shown in Fig. 3b, soot is present in both urban and rural encrustations being consistent with previous observations (Guo et al., 2010). Moreover, fly ash particles resulting from coal or oil combustion are present in all environments. Parisian samples (PA13-2 and PA14-1) show many fly ashes of a diameter size < 10 µm (primarily composed of Fe) with small gypsum crystals (few micrometers) on their surfaces (Fig. 3c). This is consistent with the catalyzer effect of combustion particles released by diesel and gasoline vehicles, which increases the rate of SO2 fixation as sulfate (Rodriguez-Navarro and Sebastian, 1996). Scarce fly ashes were also observed in samples from the city of Mantes-la-Jolie (northwest of Paris). The sample MR27-1 shows isolated halite crystals (< 10 µm, Fig. 3e), which can result from marine aerosols, in agreement with its location near the sea, and numerous fly ashes (Fig. 3d), most likely from power plants and traffic roads. The dissolution of rhombohedral calcite and subsequent precipitation of gypsum crystals is also illustrated in Fig. 3f.
In summary, the presence of particulate matter and salts highlights several local or distant sources of S-bearing compounds and a prevailing anthropogenic source in the whole Parisian basin atmosphere, which may be distinguished and quantified with the isotopic composition of sulfate.
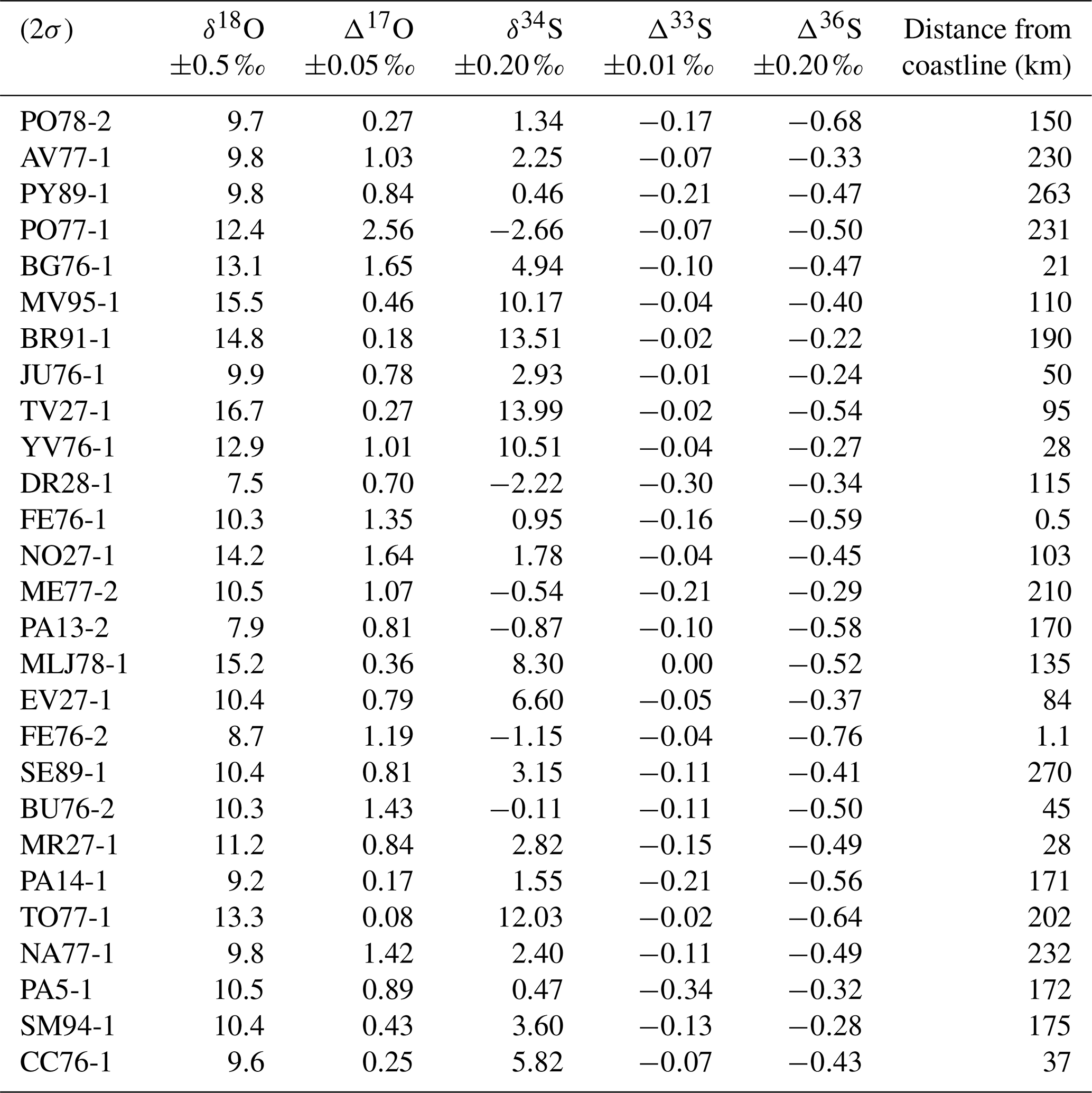
Table 2δ18O, δ34S, Δ17O, Δ33S, and Δ36S measurements of each sample with the distance from coastline.
4.2 Isotopic composition of black crust sulfates
The sulfur and oxygen isotopic compositions of black crust sulfates are reported in Table 2. The δ18O and δ34S values cover a wide range from 7.5 ‰ to 16.7 ‰ ±0.5 ‰ (2σ) and from −2.66 ‰ to 13.99 ‰ ±0.20 ‰ (2σ), respectively, with means of 11.3±2.4 ‰ and 3.78±4.79 ‰, respectively. All samples have positive Δ17O values, ranging from 0.08 to 2.56 ‰±0.05 ‰ (2σ) with an average value of 0.86 ‰. Furthermore, it is noteworthy that 67 % of black crust samples have Δ17O > 0.65 ‰. The Δ33S and Δ36S values are both negative and vary between −0.34 ‰ and 0.00±0.01 ‰ and between −0.76 ‰ and (2σ), respectively. No obvious correlation exists between δ18O, Δ17O, δ34S, and Δ33S and the distance from coastline (Fig. S1 in the Supplement).
5.1 The δ34S–δ18O–Δ17O systematics
Sulfate in black crusts may have multiple origins that could be either primary or secondary. We refer to primary sulfates here as sulfates that are not formed in the atmosphere from SO2 oxidation. They can originate from the host rock itself where sulfur occurs both as sulfide, such as pyrite that would be subsequently dissolved and oxidized as sulfate, and as carbonate-associated sulfates (CASs), which substitute for carbonate in the lattice. Sulfates in black crusts can also have been directly emitted into the atmosphere for instance by sea spay, resulting in sea-salt sulfate aerosols, or as products of combustion by refineries, vehicle exhaust, or biomass burning; these commonly correspond to “primary sulfates” in the literature. On the contrary, secondary sulfates result from the oxidation of tropospheric S-bearing gases (mainly SO2) and other compounds including dimethyl sulfide (DMS, (CH3)2S) by various oxidants (O3, H2O2, OH, O2 TMI, and NO2). As black crusts are mainly consisted of gypsum (CaSO42H2O), coupled δ34S–δ18O variations can be used to trace natural vs. anthropogenic sources of sulfates in black crusts. Constrains on the primary/secondary origins of sulfate aerosols can also be brought by Δ17O values. Indeed, large positive Δ17O anomalies in sulfate aerosols are inherited from their atmospheric oxidants that were produced during O3 photochemically induced genesis. In theory, other mechanisms exist such as the magnetic isotope effect (see Sect. 5.2.2) but have not been recognized yet. Resulting from photochemical reactions, O3 molecules possess O-MIF (oxygen mass independent fractionation) compositions with Δ17O of ∼35 ‰ (Janssen et al., 1999; Lyons, 2001; Mauersberger et al., 1999) with lower values in the troposphere of ∼26 ‰ (Vicars and Savarino, 2014). Every molecule inheriting oxygen atoms from O3 will also have positive Δ17O values, including H2O2 with an average Δ17O of ∼1.3 ‰ (Savarino and Thiemens, 1999). OH, which isotopically exchanges with water vapor, and O2 TMIs have mass-dependent compositions with Δ17O values of ∼0 (Dubey et al., 1997; Holt et al., 1981; Lyons, 2001) and ‰ (Barkan and Luz, 2005), respectively. Savarino et al. (2000) measured the O isotopic compositions of sulfates derived from these oxidation pathways and showed that OH and O2 TMI oxidation channels do not result in mass-independent fractionation signatures (Δ17O =0 ‰ and −0.09 ‰, respectively), whereas O3 and H2O2 radicals transfer one-quarter and one-half, respectively, of their isotopic anomalies to the sulfate, thus resulting in mass-independent fractionation signatures (Δ17O =8.75 ‰ and 0.65 ‰, respectively) (e.g., Bao et al., 2000, 2001a, b, 2010; Jenkins and Bao, 2006; Lee et al., 2002; Lee and Thiemens, 2001; Li et al., 2013; Martin et al., 2014). Mass-dependent isotopic fractionation during SO2 oxidation may change δ17O and δ18O but not the Δ17O that only depends on the mixing of O reservoirs with variable Δ17O. The fact that most black crusts have Δ17O > 0.6 ‰ demonstrates that a significant amount of sulfates is of atmospheric origin.
Black crust sulfates analyzed in this study have O and S isotopic compositions that overlap other black crusts from Europe (Fig. 4; Longinelli and Bartelloni, 1978; Torfs et al., 1997, Kramar et al., 2011, Vallet et al., 2006) and sulfate aerosols from the USA and China (Fig. 5; Bao et al., 2001a; Jenkins and Bao, 2006; Lee and Thiemens, 2001; Li et al., 2013; Romero and Thiemens, 2003). In particular, there is a positive correlation between δ34S and δ18O covering a large range of variation of ∼17 ‰ and ∼9 ‰, respectively (Fig. 4), which can be interpreted in two ways: either a process leads to a variable enrichment or depletion of 18O and 34S in the crusts or it reflects a mixing between at least one depleted (in both 18O and 34S) and one enriched end-member. In the following paragraphs, we discuss in detail the respective roles of several processes (e.g., partial SO2 oxidation, mixing) that could lead to this correlation and overprint (or not) of the source signatures. As with previous studies, we will conclude that δ34S–δ18O–Δ17O values do record a mixing between different (natural and anthropogenic) sources, but addressing the role of processes is important (a prerequisite) to assess the consistency of the Δ33S anomaly origin.
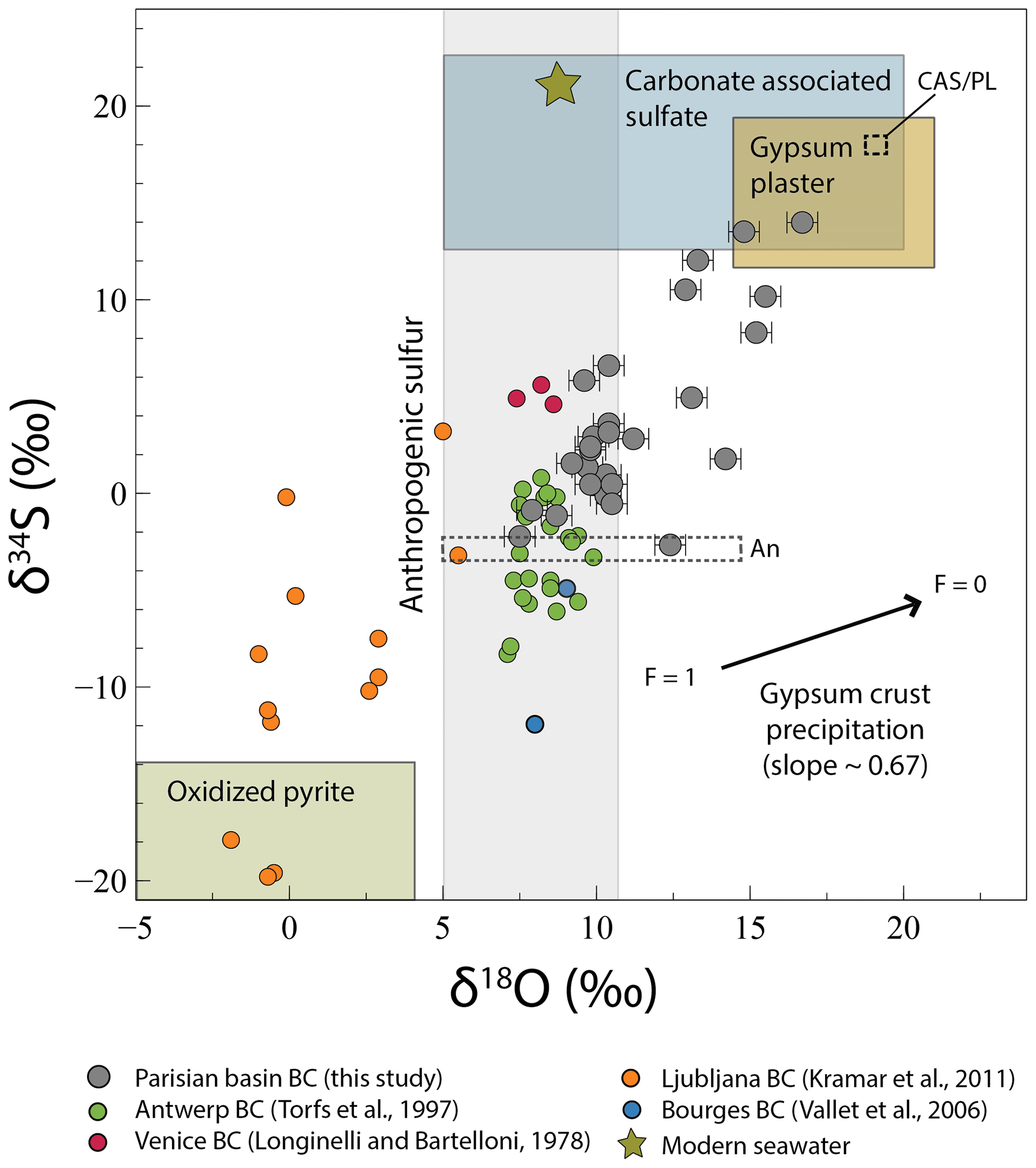
Figure 4Evolution of δ34S with δ18O in black crust (BC) sulfates. Modern seawater δ18O and δ34S are from Markovic et al. (2016) and Rees et al. (1978), respectively. The extreme anthropogenic sulfate δ18O and δ34S are from Lee et al. (2002) and Faure (1986) respectively. The carbonate-associated sulfate compositions are from Rennie and Turchyn (2014) and Turchyn et al. (2009), and those of gypsum plaster come from Kloppmann et al. (2011). Isotopic compositions determining the oxidized pyrite field are from Canfield (2004) for sulfur and are calculated following a Rayleigh distillation model with an initial H2Oδ18O =6 ‰ and a mean fractionation factor of 1.010 (Gomes and Johnston, 2017) for oxygen. The black arrow represents the fractionation induced by gypsum precipitation where F=1 means that all sulfates are dissolved and F=0 means that all sulfates are precipitated. The dashed fields represent the sulfur isotopic composition of the two anthropogenic (An) and CAS/plaster (CAS/PL) end-members.
5.1.1 Processes affecting O and S isotopic compositions
Firstly, gypsum precipitation would fractionate both O and S isotopes following a slope of 0.67±0.02 (Fig. 4) when considering fractionation factors for 18O∕16O between the dissolved sulfate and the gypsum of ∼1.002 or 1.0036 (experimental and natural values, respectively) (Lloyd, 1968). For 34S∕32S, the ranges would be between 1.000 and 1.0024 (Ault and Kulp, 1959; Nielsen, 1974; Raab and Spiro, 1991; Thode et al., 1961), and a Rayleigh-type process in which black crusts represents the cumulated (precipitated) product at different residual fraction F of dissolved sulfates that are leached. However, the slope defined by the samples is steeper, ∼1.52 (R2=0.58), implying that the gypsum precipitation is not the main mechanism driving δ34S and δ18O variations in black crusts. Another process that could affect δ34S–δ18O values is the partial oxidation of SO2 by different oxidants (e.g., O2 TMI, H2O2, O3, OH). Using the fractionation factors obtained experimentally at 19 ∘C by Harris et al. (2012a) for each oxidant ( (OH) ; (H2O2) ; (O2 TMI) ; (O3) ) and their respective proportions from Sofen et al. (2011), usually cited in the literature for present-day atmosphere (27 % OH; 18 % O2 TMI; 50 % H2O2; 5 % O3), we calculated a global fractionation factor of 1.0097. It is worth noting that these values produce a Δ17O of 0.66 ‰ in agreement with our mean Δ17O in black crusts. Consequently, following a Rayleigh distillation model and an initial SO2 with δ34S =0 ‰, the cumulated sulfates representing the black crusts would increase up to ∼9 ‰ at maximum when < 10 % SO2 is oxidized, which cannot explain the ∼17 ‰ δ34S variation, especially since 40 % oxidized SO2 is reported (Chin et al., 2000). To generate δ34S values as high as 17 ‰, O3 and H2O2 oxidation pathways should increase drastically (i.e., requiring the absence of an O2 TMI pathway), predicting an increase of Δ17O up to ∼6.5 ‰, which is not consistent with Δ17O ∼0 ‰ associated with high δ34S (Fig. 5). Therefore, SO2 partial oxidation can explain a part of the data but not the whole isotope variations. The large δ34S range could also reflect temporal variation, since in Greenland ice cores δ34S was > 10 ‰ before the industrial period (Patris et al., 2002), which is dominated by SO2 from DMS (Sofen et al., 2011) and then decreased < 4 ‰ in the 1960s, which is dominated by anthropogenic SO2. Following this variation, black crusts on recently renovated churches should display low δ34S and those renovated before the industrial period should display higher δ34S. However, samples ME77-2 (δ34S ‰) and EV27-1 (δ34S =6.60 ‰) compared to PY89-1 (δ34S =0.46 ‰) gathered on churches restored after World War II and in 1772, respectively, present no significant temporal variation, which might be due to higher proportions of anthropogenic SO2 emitted recently (0.5 Tg S yr−1 before the industrial period and up to 69 Tg S yr−1 for the present day; Sofen et al., 2011, and references therein). Thus, black crusts do not seem to record temporal isotopic variation, even if samples with δ34S ‰ and δ34S =13.99 ‰ should be dated to confirm this assumption. Alternatively, with well-exposed surfaces to precipitation emphasizing wash-out and subsequent reprecipitation, black crusts could rather probe “recent” SO2 oxidation. So far, no known processes seem to affect the isotopic compositions, which rather probe the source signatures.
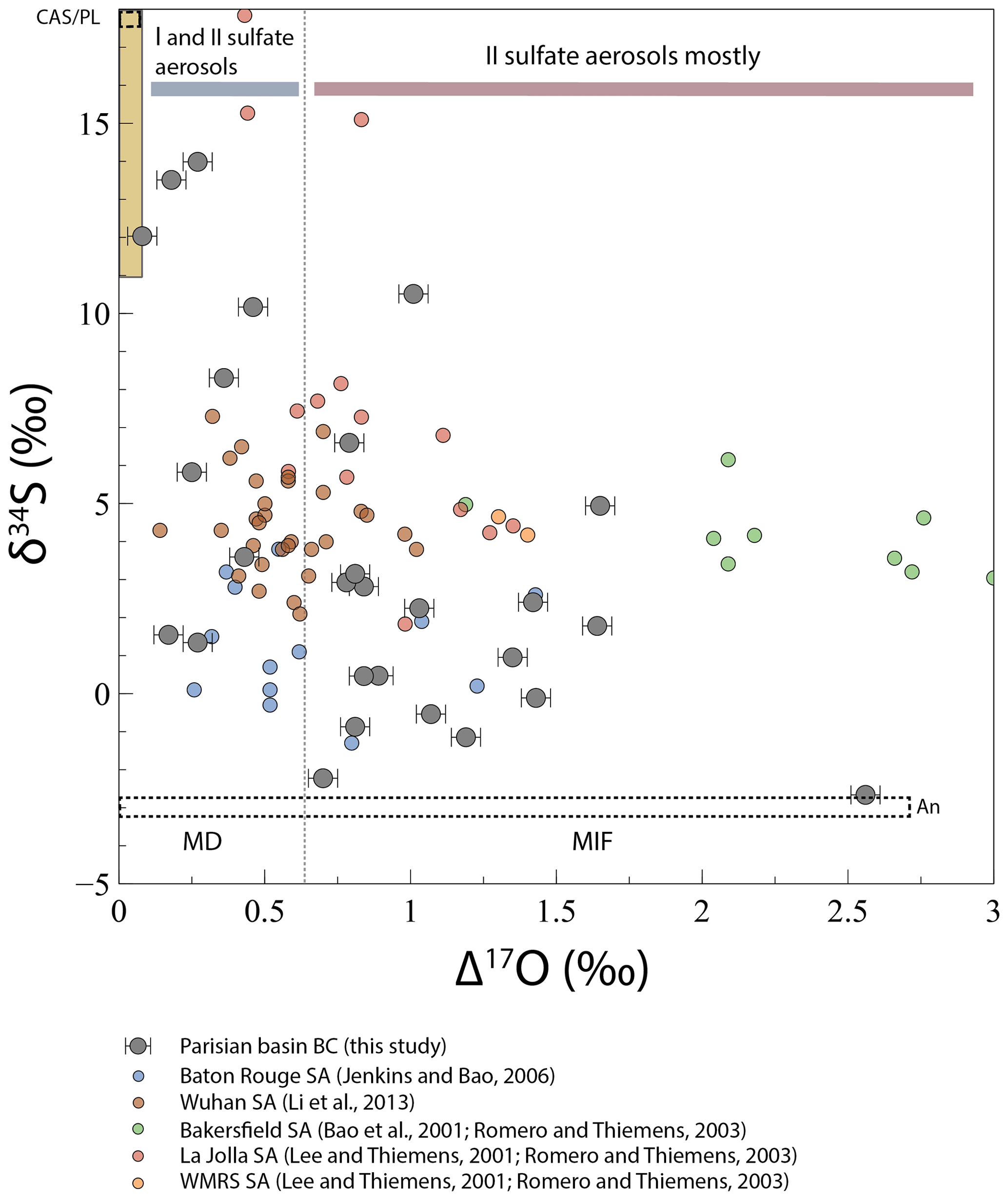
Figure 5Evolution of δ34S with Δ17O in black crust (BC) sulfates and sulfate aerosols (SA). The limit between mass-dependent and mass-independent fractionation (dashed line) is defined for Δ17O ∼0.65 ‰, where H2O2 will be the major oxidant, giving its O anomaly to sulfates (Savarino et al., 2000). When Δ17O < 0.65 ‰, black crust sulfates result from a mixing between primary sulfates (gypsum plaster and CAS and/or anthropogenic sulfur) and secondary sulfate aerosols, where SO2 is oxidized by H2O2 (Δ17O =0.65 ‰), OH (Δ17O =0 ‰), or O2 TMI (Δ17O ‰) mainly (gray bar). When Δ17O > 0.65 ‰, black crust sulfates represent secondary sulfate aerosols mainly, resulting from a mixing between SO2 oxidized by O3 (Δ17O =8.75 ‰) and H2O2 (red bar). The yellow array represents gypsum plaster and CAS isotopic compositions. The dashed fields represent the sulfur isotopic composition of the two anthropogenic (An) and CAS/plaster (CAS/PL) end-members.
5.1.2 Source effects
If δ34S–δ18O variation reflects mixing of sources, at least two end-members are required. Determined graphically in Figs. 4 and 5, a first one would be 18O–34S enriched both around 18 ‰ with a near-zero Δ17O, which in view of the sampling cross section from NW to SE and west-dominating winds could correspond to the sea-spray isotopic signature, but available data usually display δ18O of ∼9 ‰ (Markovic et al., 2016) and δ34S ∼21 ‰ (Rees et al., 1978), ruling out the occurrence of sea-sprays. With the DMS produced by phytoplankton and oxidized in the atmosphere (1125 Tg S yr−1) being higher than sea-salt emissions (6–12 Tg S yr−1 (Alexander et al., 2005, and references therein), with δ34S of 15 ‰–20 ‰ (Calhoun et al., 1991), sulfate aerosols deriving from DMS oxidation could rather represent this 18O–34S-enriched end-member. However, the absence of a correlation between δ34S, δ18O, Δ17O and the distance from coastline (Fig. S1) and near-zero Δ17O for high δ34S values (Fig. 5) is not consistent with a significant DMS contribution, mostly oxidized by O3 (see above; Alexander et al., 2012). Despite some isolated halite crystals observed in one sample (Fig. 3e), we conclude that, overall, marine aerosols (DMS, sea-salt sulfates) do not relate to the high δ34S–δ18O end-member. Major-element contents (e.g., Na, Cl) have not been measured here even if they could further constrain and quantify the presence of marine aerosols. The structural analyses of black crusts emphasize dissolution of the underlying carbonate. Carbonate-associated sulfates (with S abundances varying between a few tens to thousands of parts per million (ppm; Kampschulte and Strauss, 2004, and references therein) would also be dissolved and reprecipitated in black crusts and may well represent the enriched δ34S–δ18O end-member with near-zero Δ17O. CAS analyses from Atlantic and Pacific oceans over the last 25 Myr and in the middle Cretaceous Tethys ocean show δ34S from 11 ‰ to 24 ‰, with δ18O from 5 ‰ to 21 ‰ similar to barite isotopic composition (Rennie and Turchyn, 2014; Turchyn et al., 2009). Furthermore, marine sulfates typically have Δ17O of ∼0 and > −0.2 ‰ in the geological record (Bao et al., 2008). CAS would thus perfectly match this end-member. Plaster used to seal blocks of carbonate stones is made through Lutetian gypsum dehydration and could also well represent the 18O–34S-rich end-member. Indeed, Kloppmann et al. (2011) measured δ34S between 12.6 ‰ and 18.3 ‰ and δ18O from 14.6 ‰ to 21.5 ‰ for mortars and plasters from French churches and castles. Thus, the surrounding plaster also matches this end-member, which is named CAS/Pl in Figs. 4 and 5.
The depleted end-member is graphically characterized by δ34S < −3 ‰ with little constrained δ18O from 5 ‰ to 15 ‰ (Fig. 4, dashed box “An”) and Δ17O from ∼0 ‰ to 2.6 ‰ (Fig. 5, dashed box An). Sulfates from dissolved and oxidized sedimentary pyrites contained in the building carbonate stone are known to have δ34S < −12 ‰ (since at least the last 500 Myr; Canfield, 2004). Despite a sulfide content that can vary between a few tens to thousands of parts per million (Thomazo et al., 2018), our sampled carbonate stones are very whitish, suggesting a low sulfide content. Even if it would certainly not affect the mass balance, we took into account pyrite oxidation, as other studies did on black crusts (Kramar et al., 2011; Vallet et al., 2006). With the S isotope fractionation factor during pyrite oxidation being negligible (between 0.996 and 1; Thurston et al., 2010, and references therein) compared to O isotopes, we modeled the δ18O variation according to a Rayleigh distillation to represent the sulfide oxidation, commonly occurring via O2+H2O at the atmosphere–carbonate building stone interface. With an initial δ18O of ‰ of rainwater in the Paris Basin and a mean 18αwater-sulfate of 1.010 (Gomes and Johnston, 2017), sulfates from pyrite oxidation would have δ18O of ∼4 ‰ and as low as −6 ‰ if water would be in limited amounts (i.e., residual fraction of water F∼0). Very recently, pyrite oxidation was hypothesized to occur via O3 (Hemingway et al., 2019), which would lead to positive Δ17O of sulfates with low δ34S, explaining the depleted end-member. However, our data are strikingly higher than for black crusts from Ljubljana (Slovenia; Kramar et al., 2011), which show δ34S as low as −20 ‰ and δ18O between −2 ‰ and 5 ‰ (Fig. 4) that would be typical for pyrite oxidation. Besides, there is so far no evidence for a higher oxidation flux of pyrite via O3 than major constituents as H2O and O2. This means that another source should have negative δ34S. Anthropogenic sulfur represents ∼60 % of the total sulfur released worldwide and includes primary sulfates as oil, coal, and biomass combustion products as well as SO2 emission that can be oxidized into secondary sulfates. When considering coal and oil combustion, δ34S can vary largely between −30 ‰ and 32 ‰ (e.g., Faure, 1986). More locally, a recent study reported a narrow range from −0.57 ‰ to 11.33 ‰ for sulfur emitted by transport and industries in Paris (Au Yang et al., 2020). Lee et al. (2002) carried out vegetation and diesel combustion experiments, resulting in δ34S and δ18O values between 9.55 ‰ and 16.42 ‰ and between 5.5 and 10.5 ‰, respectively, with Δ17O of ∼0 ‰, forming primary sulfates without mass-independent signatures. Because sulfate aerosols can be either primary or secondary with various SO2 oxidation pathways having distinct δ18O–Δ17O values and O-fractionation factors, atmospheric aerosols would result in variable δ18O–Δ17O values. Therefore, the depleted end-member with δ34S < −3 ‰ and 0 < Δ17O < 0.65 ‰ could be typified by primary anthropogenic sulfate aerosols, or SO2 oxidized by OH or O2 TMI, and/or a subtle mixing of oxidation pathways to yield near-zero Δ17O, whereas samples with Δ17O > 0.65 ‰ rather point to a significant anthropogenic SO2 fraction oxidized by O3 and H2O2 or by O3 and to a lesser extent by O2 TMI and OH, depending on the water pH (Lee and Thiemens, 2001), corresponding to secondary sulfate aerosols (named An in Figs. 4 and 5). As the distinction between primary and secondary sulfate aerosols having near-zero Δ17O is not possible, we assume a mixing with only two end-members, CAS/Plaster and anthropogenic sulfur (primary and secondary). Furthermore, in view of the O isotope variability caused by the oxidation, mixing proportions were calculated based only on δ34S values. We chose the end-members graphically and in agreement with the literature, i.e., a CAS/PL δ34S value of 18 ‰, in the range from 11 ‰ to 24 ‰ (Kloppmann et al., 2011; Rennie and Turchyn, 2014; Turchyn et al., 2009) (Fig. 4) and an An δ34S value of −3 ‰, similar to Montana et al. (2008) as well the closest to sulfates measured in Paris. CAS/PL proportions range from 2 % to 81 % with an average of ∼32 %. With an extreme δ34S of −10 ‰ for the An end-member, encompassing black crusts from Antwerp, the CAS/PL proportion averaged 49 %. This highlights that host-rock sulfate is on average not the main S provider, and that black crusts record atmospheric sulfate aerosols. Excluding the most “contaminated” samples by CAS/PL and assuming that those having Δ17O > 0.65 ‰ obviously represent SO2 oxidized by O3 and H2O2, the minimum proportion of MIF-bearing sulfates, and hence secondary sulfates, can be estimated at ∼63 %, which is close to estimations by Lee and Thiemens (2001) and Sofen et al. (2011). In summary, black crusts sample significant amounts of atmospheric SO2 and complement existing sampling such as aerosols, which allow us to address the origin of the Δ33S anomaly.
5.2 Black crusts S-MIF signature
5.2.1 Processes implicated in black crust formation
Δ33S–Δ36S values recorded by black crust sulfates range from −0.34 ‰ to 0.00±0.01 ‰ for Δ33S and from −0.76 ‰ to ‰ (2σ) for Δ36S (Table 2). These values are quite unusual compared with anthropogenic and natural aerosols. As illustrated by Fig. 6, black crust sulfate Δ33S values are all negative, and it is worth noting that this depletion occurs with near-constant Δ36S values. This is somewhat distinct from most aerosols, which display almost exclusively positive Δ33S values up to ∼0.5 ‰ and both positive and negative Δ36S values (Au Yang et al., 2019; Guo et al., 2010; Lin et al., 2018b; Romero and Thiemens, 2003; Shaheen et al., 2014). So far, the only negative Δ33S values down to −0.6 ‰ were measured in sulfate aerosols from Beijing (China) during winter months (Han et al., 2017) (no Δ36S values provided), and these values were assumed to result from incomplete combustion of coal. This assumption ultimately relies on the work of Lee et al. (2002), which showed that primary anthropogenic aerosols formed by high-temperature combustion (e.g., diesel) result in near-zero Δ33S–Δ36S values, whereas those formed by low-temperature combustion (e.g., biomass burning) result in Δ33S down to −0.2 ‰ and Δ36S values varying between −1.9 ‰ and 0.2 ‰ (data recalculated with 36β=1.9). Negative Δ36S values well correlated with biomass burning proxies are also reported in East China (Lin et al., 2018b), although Δ33S was ∼0 ‰. As in many other cities, Paris has long been affected by coal and wood burning, we can hypothesize that Δ33S–Δ36S variations result from high- and/or low-temperature combustion processes. Some black crust sulfates with near-zero Δ33S-Δ36S values could result from high-temperature combustion, but this would not explain negative Δ33S–Δ36S values. Furthermore, according to Lin et al. (2018b), low-temperature combustion would preferentially fractionate 36S over 33S, which should result in a steep slope in Δ33S–Δ36S space. The trend defined by our black crust samples shows higher 33S fractionation than 36S with Δ33S values lower than those obtained by available low-temperature combustion experiments (< −0.2 ‰; Lee et al., 2002) and with Δ36S values in the range of aerosols. Furthermore, no Δ33S evolution is observed in black crusts sampled on churches with different ages of renovation (see Sect. 5.1; ME77-2 Δ33S ‰; EV27-1 Δ33S ‰; and PY89-1 Δ33S ‰), whereas we would expect a Δ33S increase in black crusts from −0.2 ‰ to 0 ‰ due to the reduction of sulfur emission from low-temperature replaced by high-temperature combustion processes. Therefore, available data highlight that neither high- nor low-temperature combustion processes are responsible for low Δ33S measured in black crusts.
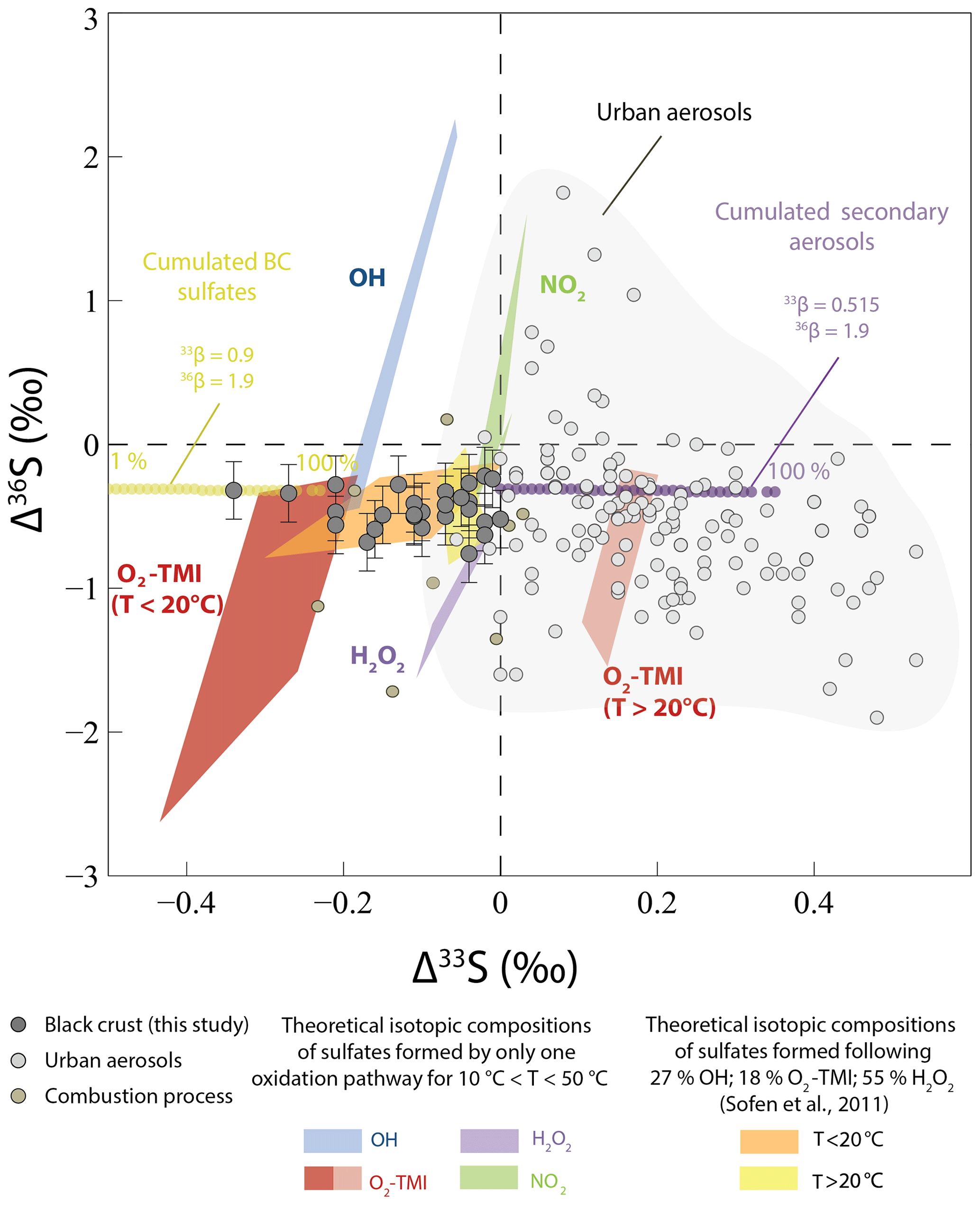
Figure 6Δ33S and Δ36S of the black crust, compared to sulfates formed by different oxidation pathways and by a mixing of them in the proportions estimated by Sofen et al. (2011). We took , 33βOH=0.503, (for T < 20 ∘C), (for T > 20 ∘C), and , 36βOH=1.97, (for T < 20 and T > 20 ∘C), and (Au Yang et al., 2018; Harris et al., 2013b). As and are unknown, we modified the proportions by Sofen et al. (2011) as follows: 27 % OH, 18 % O2 TMI, 55 % H2O2, and 0 % O3. The urban aerosol isotopic compositions are a compilation from Au Yang et al. (2019); Guo et al. (2010); Lin et al. (2018b); Romero and Thiemens (2003); and Shaheen et al. (2014), while the combustion process reflects samples from Lee et al. (2002). Modeled Δ33S–Δ36S values of cumulated black crusts (BC) sulfates – formed by SO2 wet and dry deposition with a magnetic isotope effect (MIE; 33S depletion compared to initial SO2 with constant negative Δ36S) and of cumulated secondary aerosols formed by SO2 oxidation by O3, O2 TMI, OH, and H2O2 (33S enrichment compared to initial SO2) from an initial SO2 with Δ33S–Δ36S =0 ‰ – are reported with corresponding β exponents (see Sect. 5.2.2 for model explanation). Residual SO2 and global cumulated BC plus secondary aerosol isotopic compositions were not reported for better readability. Percentages indicate the fraction of produced cumulated BC and secondary aerosols.
With a part of black crust sulfates being atmospheric in origin, isotopic effects during SO2 oxidation could be responsible for Δ33S–Δ36S variations. To better address this issue, we calculated the Δ33S–Δ36S values of sulfates predicted by each of the main SO2 oxidation pathways and by a mixing of them in the proportions given by Sofen et al. (2011). We used 33β and 36β values determined by experiments of SO2 oxidation by O2 TMI, H2O2, O3, OH (Harris et al., 2013b), and values cited in Au Yang et al. (2018) (see caption text). We also used NO2 values (Au Yang et al., 2018) and T-dependent equations determined by Harris et al. (2013b) to calculate each 34α with initial sulfur dioxide Δ33S and Δ36S of 0 ‰ (Lin et al., 2018b). As mentioned earlier (Au Yang et al., 2018; Harris et al., 2013b), none of these models can account for anomalous Δ33S–Δ36S values in either aerosols or in black crusts (Fig. 6). Although oxidation with O2 TMI at T=50 ∘C could produce negative Δ33S down to −0.37 ‰, which would account for the lowest Δ33S observed in black crusts, this oxidation pathway would also produce larger Δ36S down to −1.50 ‰ at odds with the Δ36S reported in the black crust. Their potential combination cannot account for sulfate aerosol data from the literature (Au Yang et al., 2019; Guo et al., 2010; Lin et al., 2018b; Romero and Thiemens, 2003; Shaheen et al., 2014) or for the black crust as it would result in slightly negative Δ33S–Δ36S values that could not explain the Δ33S as low as −0.34 ‰ (yellow frames in Fig. 6). Available literature data are therefore not consistent with the anomalous Δ33S–Δ36S values recorded in black crust sulfates.
Mass-dependent processes can also result in small Δ33S–Δ36S variations, depending on the magnitude of the 34S fractionation (Ono et al., 2006a). As mentioned in Sect. 5.1.2, a mixing between a 33,34S-depleted end-member (An) consisting of anthropogenic sulfur (δ34S ‰, Δ33S =0 ‰) and a 33,34S-enriched sulfates end-member (CAS/PL) from plaster or CAS (δ34S =18 ‰, Δ33S =0 ‰) would result in small Δ33S values of −0.01 ‰ for 50 % mixing, which is far from the maximum measured Δ33S of . Moreover, the slope between Δ33S–Δ36S would be about −7, which is at odds with our observations. Therefore, we conclude that mixing cannot account for the black crust Δ33S–Δ36S variations.
5.2.2 A new oxidation pathway implying magnetic isotope effect
Several studies proposed that positive Δ33S measured in sulfate aerosols, with Δ33S up to 0.5 ‰, from, for example, East China and California could result from stratospheric fallout of SO2 (with Δ33S potentially up to 10 ‰ higher; Ono et al., 2013), which underwent UV photolysis by short-wavelength radiation (Romero and Thiemens, 2003; Lin et al., 2018a, b). This suggestion primarily relies on the similarities between Δ33S–Δ36S values of sulfate aerosols and laboratory experiments of SO2 photolysis conducted at different wavelengths (Romero and Thiemens, 2003) and on the correlation between 35S-specific activity and Δ33S values (Lin et al., 2018b). However, these studies never addressed the absence of the complementary negative Δ33S reservoir, which is required to balance the positive Δ33S reservoir (see Au Yang et al., 2019). In this respect, it is worth mentioning that volcanic and stratospheric aerosols trapped in Antarctic ice cores (see Gautier et al., 2018, and references therein) show both positive Δ33S (up to ∼2 ‰) and complementary negative Δ33S values (down to −1 ‰) and weighed average Δ33S ≠0 ‰ explained by prior partial deposition. Stratospheric fluxes are actually too low to account for Δ33S > 0.1 ‰ (Lin et al., 2016; Au Yang et al., 2019). Accordingly, some other authors rather tried to explain the positive anomalies of most aerosols with “tropospheric” chemical reactions, which are SO2 oxidation by the main oxidant including NO2, H2O2, OH, O3, and O2 TMI, but experimental data result in a maximum Δ33S values of ∼0.2 ‰ for all studied reactions (Au Yang et al., 2018; Harris et al., 2013b). Isotope effects associated with SO2 oxidation by minor species, such as Criegee radicals, remains to be investigated (Au Yang et al., 2018). In summary, whatever the stratospheric vs. tropospheric origin of positive Δ33S values recorded by most aerosols, there is a 33S isotope imbalance and a missing reservoir with negative Δ33S that must exist. Han et al. (2017) reported Δ33S values down to −0.6 ‰ in sulfate aerosols from Beijing. As discussed above, the authors' suggestion calling for low-temperature combustion is little supported by available data, and clearly the very restricted location and time interval, over a month, where these anomalies occurred cannot counterbalance, both spatially and temporally, the common positive Δ33S values of most aerosols; the missing reaction or reservoir requires, instead, to be ubiquitous worldwide.
In this study, black crust sulfates display negative Δ33S values (from ∼0 ‰ down to −0.34 ‰). These values are certainly produced by tropospheric chemical reactions. Otherwise, they would have, according to the stratospheric origin model, the same sign as those measured among aerosols. Furthermore, the chemical reactions (or reaction) involved in the formation of black crusts must be distinct compared to those leading to the formation of tropospheric aerosols. As developed thoroughly, black crusts could well represent the missing sulfur reservoir.
An additional observation is that negative Δ33S values occur with near-constant Δ36S (from −0.76 ‰ to ‰; Fig. 6). This signature is typical of the magnetic isotope effect (MIE), which involve a radical pair, where coupling between the nuclear magnetic moment of the nucleus of odd isotopes and the electron occurs, allowing for electron spin transition from singlet to triplet (or vice versa) (Buchachenko et al., 1976). This leads to distinct half-lives between odd and even isotopes, resulting in specific odd over even isotope enrichment (or depletion). MIE has been so far reported for various reactions occurring on a surface (Buchachenko, 2001, 2000; Turro, 1983) such as sulfate thermochemical reduction (Oduro et al., 2011) or Fe reduction in magneto-tactic bacteria (Amor et al., 2016), which are the most geologically relevant. It is worth pointing out that MIE could also be responsible for positive Δ17O values measured in black crusts, i.e., as opposed to the Δ17O anomaly being inherited from SO2 oxidants. However, Lee et al. (2002) also measured the O multi-isotope compositions of sulfate aerosols (i.e., from the atmosphere as opposed to reaction on a solid substrate) from Paris and obtained Δ17O =0.2 and 0.8 ‰ for the Paris highway and in the 13th zone, respectively, which is in good agreement with our three samples collected in Paris (from 0.17 ‰ to 0.89 ‰). Thus, this is consistent with black crust formation recording mostly an atmospheric signal and no significant magnetic isotope effect on Δ17O.
The magnetic effect could occur on a surface such as on mineral dust suspended in the atmosphere during aerosol formation, leading to residual 33S-depleted atmospheric SO2 from which black crusts would subsequently form. This model would, however, predict that some sulfate aerosols subsequently formed to display negative Δ33S values: such values are extremely uncommon, being primarily restricted to the Beijing winter months (Han et al., 2017). Instead, the magnetic effect could occur during black crust formation, on the carbonate building stone, leading to residual 33S-enriched atmospheric SO2 from which tropospheric aerosols would subsequently be formed, which is consistent with available observations. This model would, however, predict some black crust subsequently formed to display positive Δ33S: such values have not been found yet and this may well reflect sample bias, with our data being the first reported for such samples. Both scenarios imply nonzero Δ33S values of residual atmospheric SO2, which contrast with the data by Lin et al. (2018b) showing Δ33S of ∼0 ‰ (n=5, Δ33S varying from −0.04 ‰ to 0.01±0.01 ‰). Given that, in the study of Lin et al. (2018b), SO2 was sampled close to the third largest Chinese megacity, such nonzero Δ33S values may thus be rather symptomatic of emitted (i.e., anthropogenic) SO2 rather than residual or background (i.e., after significant black crust and aerosols formation). SO2 in the Paris Basin still has to be measured to confirm this assumption, but, so far, this could be consistent with the interpretation that nonzero Δ33S values of residual or background atmospheric SO2 are erased by anthropogenic SO2 having zero Δ33S values (Au Yang et al., 2019) moving towards the local source(s) of anthropogenic SO2.
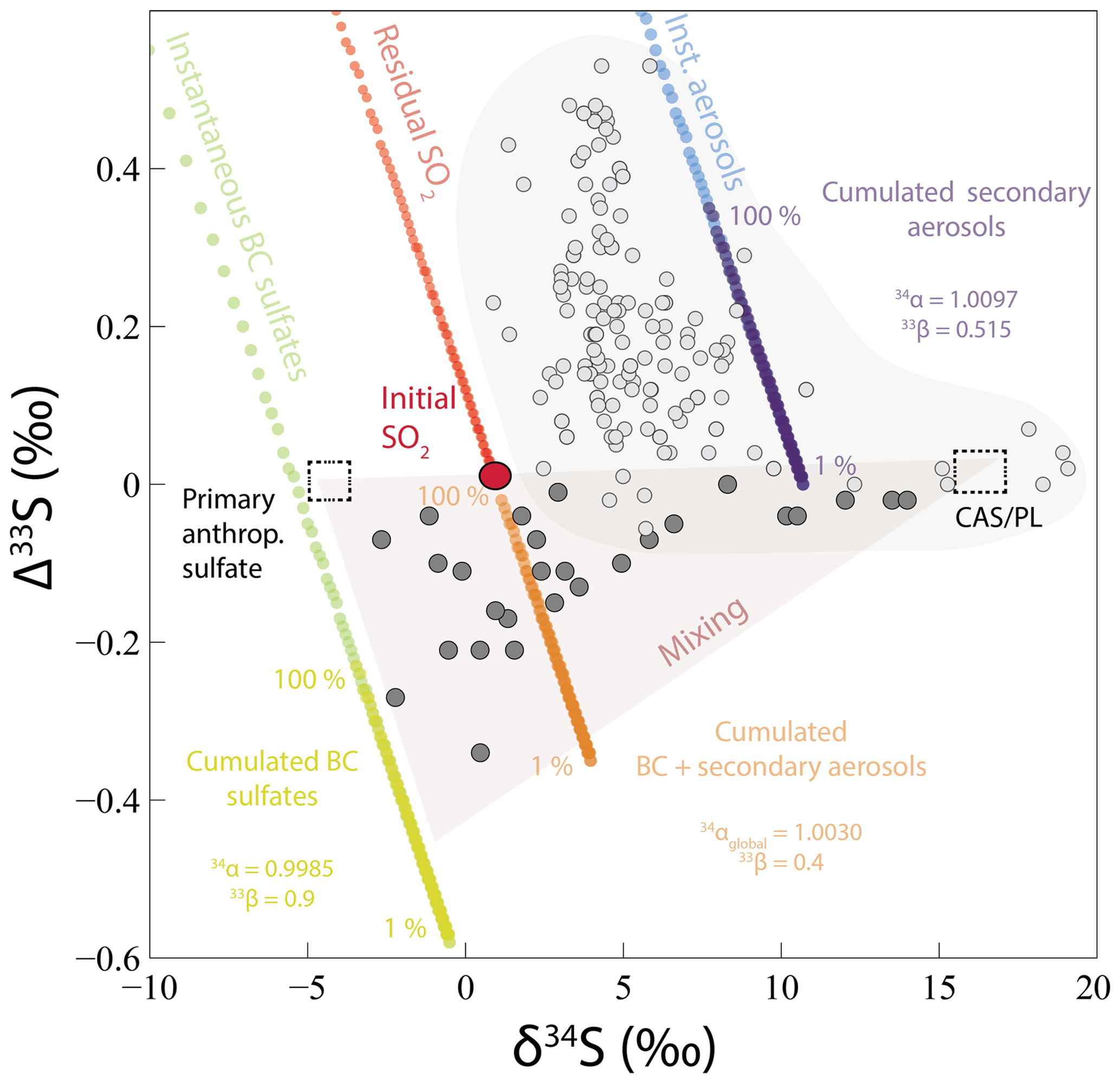
Figure 7Modeled δ34S and Δ33S values of black crust (BC) sulfates (instantaneous and cumulated) formed by SO2 wet and dry deposition with a MIE (33S depletion compared to initial SO2) and of secondary aerosols (instantaneous and cumulated) formed by SO2 oxidation by O3, O2 TMI, OH, and H2O2 (33S enrichment compared to initial SO2) from an initial SO2 with δ34S =1 ‰ and Δ33S =0 ‰ (red point). 33S enrichment of residual SO2 and global cumulated BC plus secondary aerosol isotopic compositions are also reported. Percentages indicate the fraction of produced cumulated BC and secondary aerosols (see Sect. 5.2.2 for model explanation). Black crust sulfate isotopic compositions (dark gray points) can be explained by a mixing (red triangle) between sulfates from CAS/plaster (dashed square, δ34S =18 ‰ and Δ33S =0 ‰; see Sect. 5.1), primary anthropogenic sulfates (dashed square, δ34S ‰ and Δ33S =0 ‰), and sulfates formed by wet and dry deposition of SO2 undergoing a MIE and oxidized SO2 forming secondary aerosols (cumulated BC sulfates and cumulated BC + secondary aerosols). Urban aerosol (light gray points) isotopic compositions are a compilation from Au Yang et al. (2019); Guo et al. (2010); Lin et al. (2018b); Romero and Thiemens (2003); and Shaheen et al. (2014).
In the absence of additional observations, proposing a chemical reaction, and hence a radical pair that breaks and recombines, would be very speculative, but our data clearly point towards the occurrence of a magnetic effect during the formation of black crusts, involving ubiquitous heterogeneous chemical reactions. This is supported by previous recognition of sulfur radicals such as (Herrmann, 2003) or S–S (see Babikov, 2017. But note that their Δ36S∕Δ33S slope is distinct from ours). Clearly, the reaction does not occur after sulfate formation such as during dissolution–precipitation mechanisms, which do not involve any radical species. As mentioned above, magneto-tactic bacteria can produce MIE when reducing Fe (Amor et al., 2016). With microbial activity being sometimes present on black crusts (Gaylarde et al., 2007; Sáiz-Jiménez, 1995; Scheerer et al., 2009; Schiavon, 2002; Tiano, 2002), the involvement of microorganisms, affecting only the sulfur isotopes as the most negative Δ33S, does not correspond to the most negative Δ17O, which represents another possibility to investigate. Another implication that can be tested in future work is that the kinetics of heterogeneous reactions leading to sulfate and black crust formation should be comparable or faster than those leading to aerosol formation. So far, Li et al. (2006) showed comparable loss of atmospheric SO2 by heterogeneous oxidation on calcium carbonate substrates and by gas-phase oxidation. Our conclusions show a strong analogy with the model of Au Yang et al. (2019), who suggest that SO2 photooxidation on mineral dust could form sulfate aerosols depleted in 33S that would then be deposited. The residual SO2 would be subsequently enriched in 33S, then be oxidized by common O3, H2O2, O2, or OH oxidants. Their 33S-depletion mechanism was not further constrained, except that it was speculated to be photochemical in origin.
If correct, this view requires reassessing the overall S isotope fractionation during SO2 atmospheric reaction. So far, previous studies assumed that the overall sulfur isotopic fractionation between the wet and dry deposit and oxidized SO2 was equal to 1 (i.e., no isotope effect), but negative Δ33S in black crusts is inconsistent with such an assumption.
Starting with an SO2 Δ33S value of 0 ‰ (Au Yang et al., 2018; Lin et al., 2018b) and forming oxidized (sampled by secondary aerosols) and wet and dry deposit (sampled by black crusts) reservoirs with Δ33S values up to 0.50 ‰ down to −0.34 ‰, respectively, mass balance implies that SO2 dry and wet depositions and secondary sulfate aerosols represent ∼60 % and 40 %, respectively. This is in good agreement with proportions obtained by Chin et al. (2000) and quoted by Harris et al. (2013b). Therefore, we conclude that MIE happening during SO2 dry and wet depositions could be a viable mechanism responsible for 33S enrichment of secondary sulfate aerosols and that black crusts could represent the 33S-negative complementary reservoir.
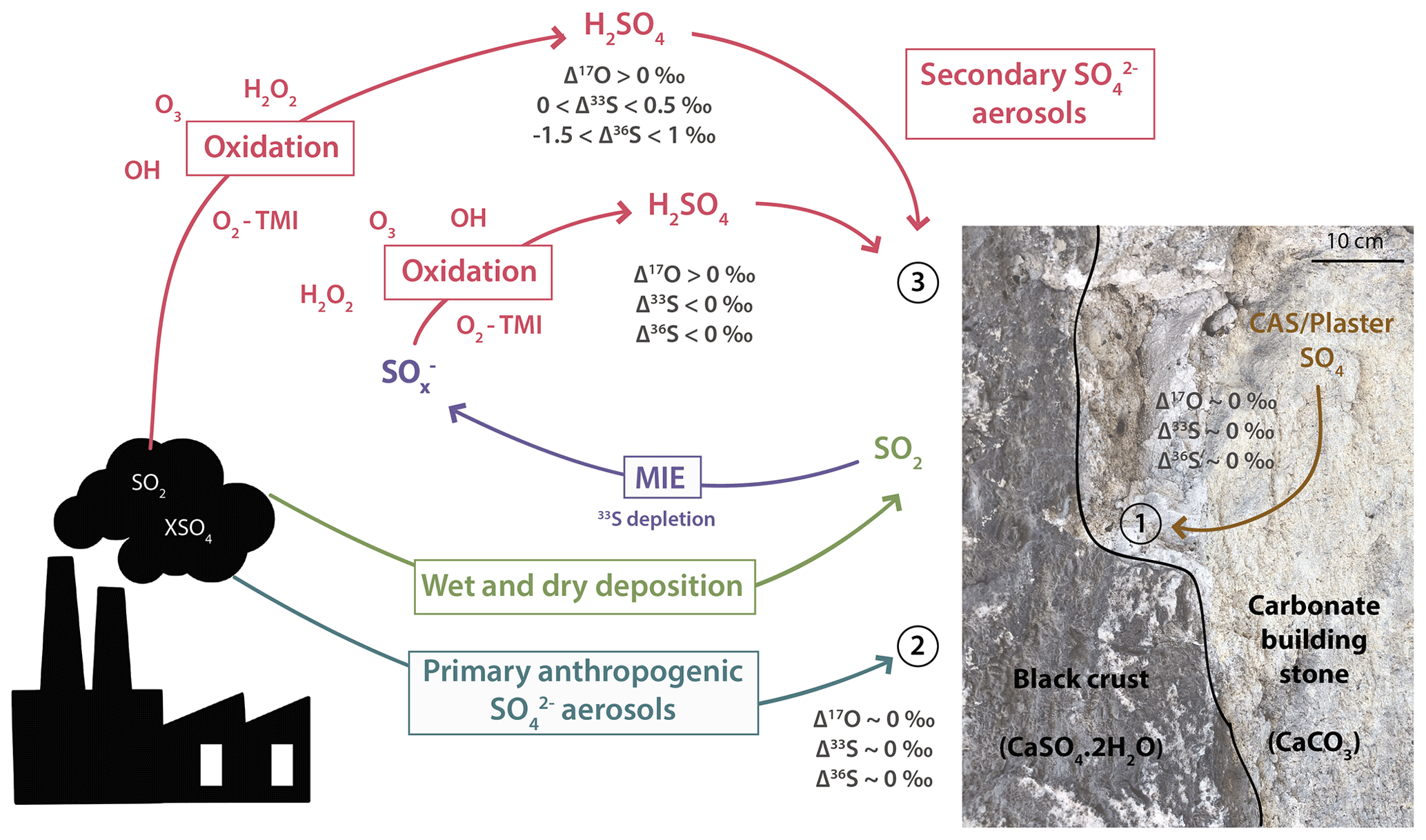
Figure 8Scheme summarizing the sulfur sources and processes that lead to black crust formation. Sulfur dioxide releases by anthropogenic activities can either be oxidized in the atmosphere by H2O2, O3, OH, O2 TMI and formed secondary sulfate aerosols that will react with the carbonate building stone to produce 33S-enriched black crust sulfates or be deposited, as dry and wet deposit, on the carbonate substrate where its oxidation into then sulfates through MIE will produce 33S-depleted black crust sulfates and a 33S-enriched residual SO2 (source 3). Primary sulfates emitted by anthropogenic activities (source 2) or carbonate-associated sulfates and/or plaster of the host rock (source 1) are also likely sources contributing to black crust formation.
In order to better apprehend the aerosols and black crust complementarity, we modeled the S isotopic fractionation of both black crusts and aerosols during SO2 oxidation (Figs. 6 and 7). We assumed a Rayleigh distillation model to represent the atmosphere–building-stone interface open system. The global fractionation factor between residual SO2 and oxidized (secondary aerosols) plus deposited (black crusts) SO2 is defined as
with A and B being the proportions of SO2 deposited and oxidized, being equal to 60 % and 40 %, respectively. This allows us to deduce the and the associated 33,36β factors. A δ34S of 1 ‰ for the initial SO2 was considered to obtain black crusts of at least −3 ‰ (see Sect. 5.1.2) and Δ33S–Δ36S =0 ‰; was taken as 1.0097 as calculated using the different oxidation channel proportions of Sofen et al. (2011). With the oxidation being mass dependent, we chose values of 0.515 and 1.9, respectively (Harris et al., 2012b). The best fit is obtained for , , and with and 1.9, respectively. The 33S enrichment in secondary sulfate aerosols is well represented by this parameterization (instantaneous and cumulated products; Figs. 6 and 7). The concomitant 33S depletion in modeled cumulated deposit is also well represented. The 33S isotopic fractionation occurring during the MIE is higher than the one observed in black crusts. To a first-order approximation, this model works, predicting the total cumulated products of black crusts and aerosols to have Δ33S values of −0.23 ‰ and 0.35 ‰, respectively. The match is not perfect, which does not entirely capture the black crust isotopic compositions. But remember that black crusts are produced from anthropogenic Parisian SO2, whereas aerosols formed in other locations possibly formed from distinct anthropogenic SO2 δ34S values. In addition, we are aware that our model strongly depends on oxidation pathways estimated by Sofen et al. (2011), which vary spatially and temporally. The main weakness is the poorly constrained estimate of intrinsic S-bearing compounds (CAS/plaster end-member) in the host rock as well as sulfate aerosols (Δ33S > 0 ‰), which dilutes the 33S anomaly, lowering the overall black crust Δ33S. Ultimately, black crusts result mainly from the deposition followed by oxidation of SO2 on the building stone rather than aerosol accumulation. The δ34S value of initial anthropogenic SO2 is another poorly constrained parameter whose variability might be difficult to estimate both spatially and temporally.
In conclusion, black crusts could represent the complementary sulfur end-member to sulfate aerosols. Its fractionation factor is relatively restricted (−1.5 ‰) and is thus likely identifiable from its negative Δ33S values. Our model is actually consistent with the assumption that the global SO2 oxidation occurs with little fractionation (34αglobal=1.00298) as commonly done in the literature. Finally, Fig. 8 summarizes the different sulfur sources involved in the black crusts as well as the processes leading to their formation. Black crust isotopic compositions could thus be explained by a mixing between sulfates from CAS/plaster (δ34S ∼18 ‰ and Δ33S =0 ‰; see Sect. 5.1 and 1 in Fig. 8), primary anthropogenic sulfates (δ34S ‰ and Δ33S =0 ‰; see 2 in Fig. 8), and wet and dry deposition of SO2 undergoing MIE during its oxidation on the building stone combined with secondary aerosols (see red triangle in Fig. 7 and process 3 in Fig. 8).
Our study shows that black crusts do preserve an atmospheric signal of SO2 oxidation, inferred from the nonzero Δ17O. Part of the sulfate originates from the surrounding plaster and/or from the stone itself, but overall > 60 % originates from anthropogenic activities. We also discovered negative Δ33S values with near-constant Δ36S signatures, which probably reflect the magnetic isotope effect involving a new oxidation pathway. The magnetic isotope effect is supposed to occur during the deposition of SO2 on the building stone surface (most likely carbonate), where SO2 is oxidized to sulfate, leading to a 33S depletion in black crust sulfates. Therefore, the resulting 33S enrichment of residual SO2 could account for positive Δ33S values of sulfate aerosols observed worldwide, making black crust sulfates their complementary Δ33S reservoir.
All data needed to draw the conclusions in the present study are shown in Table 2 and/or the Supplement. For additional data related to this study, please contact the corresponding author (genot@ipgp.fr).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-4255-2020-supplement.
IG conducted oxygen isotope measurements under the supervision of EM and ELG at IPGP. DAY conducted sulfur isotope measurements at McGill University. IG and EM collected the samples. IG, PC, EM, and DAY interpreted the data. IG wrote the paper with contributions from all coauthors. EM and MR conceived the project.
The authors declare that they have no conflict of interest.
We thank the editor Eliza Harris, Mang Lin, and an anonymous reviewer for their insightful comments as well as Mark Thiemens for his remarks on MIE that helped improve our article. We thank David Whiteley and Rémi Leprêtre for their help during the sampling, Mattauer Genot for inspiration, Omar Boudouma for the SEM analysis, Alain Person for the XRD analysis, Boswell Wing for accessing his laboratory analyses, Hao Thi Bui for assistance during analyses performed in Montreal, and Nelly Assayag and Guillaume Landais for analyses performed at IPGP. This is IPGP contribution number 4122.
This research has been supported by the ANR (grant no. 14-CE33-0009-02-FOFAMIFS).
This paper was edited by Eliza Harris and reviewed by Mang Lin and one anonymous referee.
Alexander, B., Park, R. J., Jacob, D. J., Li, Q., Yantosca, R. M., Savarino, J., Lee, C., and Thiemens, M.: Sulfate formation in sea-salt aerosols: Constraints from oxygen isotopes, J. Geophys. Res.-Atmos., 110, https://doi.org/10.1029/2004JD005659, 2005.
Alexander, B., Allman, D., Amos, H., Fairlie, T., Dachs, J., Hegg, D. A., and Sletten, R. S.: Isotopic constraints on the formation pathways of sulfate aerosol in the marine boundary layer of the subtropical northeast Atlantic Ocean, J. Geophys. Res.-Atmos., 117, https://doi.org/10.1029/2011JD016773, 2012.
Amor, M., Busigny, V., Louvat, P., Gélabert, A., Cartigny, P., Durand-Dubief, M., Ona-Nguema, G., Alphandéry, E., Chebbi, I., and Guyot, F.: Mass-dependent and-independent signature of Fe isotopes in magnetotactic bacteria, Science, 352, 705–708, 2016.
Ault, W. U. and Kulp, J.: Isotopic geochemistry of sulphur, Geochim. Cosmochim. Ac., 16, 201–235, 1959.
Au Yang, D., Landais, G., Assayag, N., Widory, D., and Cartigny, P.: Improved analysis of micro-and nanomole-scale sulfur multi-isotope compositions by gas source isotope ratio mass spectrometry, Rapid Commun. Mass Spectrom., 30, 897–907, 2016.
Au Yang, D., Bardoux, G., Assayag, N., Laskar, C., Widory, D., and Cartigny, P.: Atmospheric SO2 oxidation by NO2 plays no role in the mass independent sulfur isotope fractionation of urban aerosols, Atmos. Environ., 193, 109–117, 2018.
Au Yang, D., Cartigny, P., Desboeufs, K., and Widory, D.: Seasonality in the Δ33S measured in urban aerosols highlights an additional oxidation pathway for atmospheric SO2, Atmos. Chem. Phys., 19, 3779–3796, https://doi.org/10.5194/acp-19-3779-2019, 2019.
Au Yang, D., Widory, D., and Cartigny, P.: Coal combustion does not produce negative Δ33S, J. Geophys. Res.-Atmos., in preparation, 2020.
Babikov, D.: Recombination reactions as a possible mechanism of mass-independent fractionation of sulfur isotopes in the Archean atmosphere of Earth, P. Natl. Acad. Sci. USA, 114, 3062–3067, 2017.
Bao, H.: Purifying barite for oxygen isotope measurement by dissolution and reprecipitation in a chelating solution, Anal. Chem., 78, 304–309, 2006.
Bao, H. and Thiemens, M. H.: Generation of O2 from BaSO4 Using a Laser Fluorination System for Simultaneous Analysis of δ18O and δ17O, Anal. Chem., 72, 4029–4032, 2000.
Bao, H., Thiemens, M. H., Farquhar, J., Campbell, D. A., Lee, C. C.-W., Heine, K., and Loope, D. B.: Anomalous 17O compositions in massive sulphate deposits on the Earth, Nature, 406, 176–178, 2000.
Bao, H., Michalski, G. M., and Thiemens, M. H.: Sulfate oxygen-17 anomalies in desert varnishes, Geochim. Cosmochim. Ac., 65, 2029–2036, 2001a.
Bao, H., Thiemens, M. H., and Heine, K.: Oxygen-17 excesses of the Central Namib gypcretes: spatial distribution, Earth Planet. Sc. Lett., 192, 125–135, 2001b.
Bao, H., Lyons, J., and Zhou, C.: Triple oxygen isotope evidence for elevated CO2 levels after a Neoproterozoic glaciation, Nature, 453, 504–506, 2008.
Bao, H., Yu, S., and Tong, D. Q.: Massive volcanic SO2 oxidation and sulphate aerosol deposition in Cenozoic North America, Nature, 465, 909–912, 2010.
Barkan, E. and Luz, B.: High precision measurements of 17O∕16O and 18O∕16O ratios in H2O, Rapid Commun. Mass Spectrom., 19, 3737–3742, 2005.
Bigeleisen, J. and Mayer, M. G.: Calculation of equilibrium constants for isotopic exchange reactions, J. Chem. Phys., 15, 261–267, 1947.
Bindeman, I., Eiler, J., Wing, B., and Farquhar, J.: Rare sulfur and triple oxygen isotope geochemistry of volcanogenic sulfate aerosols, Geochim. Cosmochim. Ac., 71, 2326–2343, 2007.
Buchachenko, A., Galimov, E., Ershov, V., Nikiforov, G., and Pershin, A.: Isotopic enrichment induced by magnetic-interactions in chemical-reactions, Dokl. Akad. Nauk Sssr, 228, 379–381, 1976.
Buchachenko, A. L.: Recent advances in spin chemistry, Pure Appl. Chem., 72, 2243–2258, 2000.
Buchachenko, A. L.: Magnetic isotope effect: Nuclear spin control of chemical reactions, J. Phys. Chem. A, 105, 9995–10011, 2001.
Cabral, R. A., Jackson, M. G., Rose-Koga, E. F., Koga, K. T., Whitehouse, M. J., Antonelli, M. A., Farquhar, J., Day, J. M., and Hauri, E. H.: Anomalous sulphur isotopes in plume lavas reveal deep mantle storage of Archaean crust, Nature, 496, 490–493, 2013.
Calhoun, J. A., Bates, T. S., and Charlson, R. J.: Sulfur isotope measurements of submicrometer sulfate aerosol particles over the Pacific Ocean, Geophys. Res. Lett., 18, 1877–1880, 1991.
Camuffo, D.: Physical weathering of stones, Sci. Total Environ., 167, 1–14, 1995.
Canfield, D. E.: The evolution of the Earth surface sulfur reservoir, Am. J. Sci., 304, 839–861, 2004.
Chin, M., Rood, R. B., Lin, S. J., Müller, J. F., and Thompson, A. M.: Atmospheric sulfur cycle simulated in the global model GOCART: Model description and global properties, J. Geophys. Res.-Atmos., 105, 24671–24687, 2000.
Cowie, B. R. and Johnston, D. T.: High-precision measurement and standard calibration of triple oxygen isotopic compositions (δ18O, ) of sulfate by F2 laser fluorination, Chem. Geol., 440, 50–59, 2016.
Dauphas, N. and Schauble, E. A.: Mass fractionation laws, mass-independent effects, and isotopic anomalies, Annu. Rev. Earth Pl. Sc., 44, 709–783, 2016.
Defouilloy, C., Cartigny, P., Assayag, N., Moynier, F., and Barrat, J.-A.: High-precision sulfur isotope composition of enstatite meteorites and implications of the formation and evolution of their parent bodies, Geochim. Cosmochim. Ac., 172, 393–409, 2016.
Delavault, H., Chauvel, C., Thomassot, E., Devey, C. W., and Dazas, B.: Sulfur and lead isotopic evidence of relic Archean sediments in the Pitcairn mantle plume, P. Natl. Acad. Sci. USA, 113, 12952–12956, 2016.
Dubey, M. K., Mohrschladt, R., Donahue, N. M., and Anderson, J. G.: Isotope specific kinetics of hydroxyl radical (OH) with water (H2O): Testing models of reactivity and atmospheric fractionation, J. Phys. Chem. A, 101, 1494–1500, 1997.
Farquhar, J. and Wing, B. A.: Multiple sulfur isotopes and the evolution of the atmosphere, Earth Planet. Sc. Lett., 213, 1–13, 2003.
Farquhar, J., Bao, H., and Thiemens, M.: Atmospheric influence of Earth's earliest sulfur cycle, Science, 289, 756–758, 2000.
Farquhar, J., Savarino, J., Airieau, S., and Thiemens, M. H.: Observation of wavelength-sensitive mass-independent sulfur isotope effects during SO2 photolysis: Implications for the early atmosphere, J. Geophys. Res.-Planet., 106, 32829–32839, 2001.
Farquhar, J., Wing, B., McKeegan, K., Harris, J., Cartigny, P., and Thiemens, M.: Mass-independent sulfur of inclusions in diamond and sulfur recycling on early Earth, Science, 298, 2369–2372, 2002.
Farquhar, J., Johnston, D. T., and Wing, B. A.: Implications of conservation of mass effects on mass-dependent isotope fractionations: influence of network structure on sulfur isotope phase space of dissimilatory sulfate reduction, Geochim. Cosmochim. Ac., 71, 5862–5875, 2007a.
Farquhar, J., Peters, M., Johnston, D. T., Strauss, H., Masterson, A., Wiechert, U., and Kaufman, A. J.: Isotopic evidence for Mesoarchaean anoxia and changing atmospheric sulphur chemistry, Nature, 449, 706–709, 2007b.
Faure, G.: Isotope systematics in two-component mixtures, John Wiley and Sons, 1986.
Forster, P., Ramaswamy, V., Artaxo, P., Berntsen, T., Betts, R., Fahey, D. W., Haywood, J., Lean, J., Lowe, D. C., and Myhre, G.: Changes in atmospheric constituents and in radiative forcing, Chapter 2, in: Climate Change 2007, The Physical Science Basis, 2007.
Fronteau, G., Schneider-Thomachot, C., Chopin, E., Barbin, V., Mouze, D., and Pascal, A.: Black-crust growth and interaction with underlying limestone microfacies, Geological Society, London, Special Publications, 333, 25–34, 2010.
Gautier, E., Savarino, J., Erbland, J., and Farquhar, J.: SO2 Oxidation Kinetics Leave a Consistent Isotopic Imprint on Volcanic Ice Core Sulfate, J. Geophys. Res.-Atmos., 123, 9801–9812, 2018.
Gaylarde, C. C., Ortega-Morales, B. O., and Bartolo-Perez, P.: Biogenic black crusts on buildings in unpolluted environments, Curr. Microbiol., 54, 162–166, 2007.
Geng, L., Savarino, J., Caillon, N., Gautier, E., Farquhar, J., Dottin III, J., Magalhães, N., Hattori, S., Ishino, S., and Yoshida, N.: Intercomparison measurements of two 33 S-enriched sulfur isotope standards, J. Anal. Atom. Spectrom., 34, 1263–1271, 2019.
Gomes, M. L. and Johnston, D. T.: Oxygen and sulfur isotopes in sulfate in modern euxinic systems with implications for evaluating the extent of euxinia in ancient oceans, Geochim. Cosmochim. Ac., 205, 331–359, 2017.
Guo, Z., Li, Z., Farquhar, J., Kaufman, A. J., Wu, N., Li, C., Dickerson, R. R., and Wang, P.: Identification of sources and formation processes of atmospheric sulfate by sulfur isotope and scanning electron microscope measurements, J. Geophys. Res.-Atmos., 115, https://doi.org/10.1029/2009JD012893, 2010.
Han, X., Guo, Q., Strauss, H., Liu, C., Hu, J., Guo, Z., Wei, R., Peters, M., Tian, L., and Kong, J.: Multiple sulfur isotope constraints on sources and formation processes of sulfate in Beijing PM2.5 aerosol, Environ. Sci. Technol., 51, 7794–7803, 2017.
Harris, E., Sinha, B., Foley, S., Crowley, J. N., Borrmann, S., and Hoppe, P.: Sulfur isotope fractionation during heterogeneous oxidation of SO2 on mineral dust, Atmos. Chem. Phys., 12, 4867–4884, https://doi.org/10.5194/acp-12-4867-2012, 2012a.
Harris, E., Sinha, B., Hoppe, P., Crowley, J. N., Ono, S., and Foley, S.: Sulfur isotope fractionation during oxidation of sulfur dioxide: gas-phase oxidation by OH radicals and aqueous oxidation by H2O2, O3 and iron catalysis, Atmos. Chem. Phys., 12, 407–423, https://doi.org/10.5194/acp-12-407-2012, 2012b.
Harris, E., Sinha, B., Van Pinxteren, D., Tilgner, A., Fomba, K. W., Schneider, J., Roth, A., Gnauk, T., Fahlbusch, B., and Mertes, S.: Enhanced role of transition metal ion catalysis during in-cloud oxidation of SO2, Science, 340, 727–730, 2013a.
Harris, E., Sinha, B. R., Hoppe, P., and Ono, S.: High-precision measurements of 33S and 34S fractionation during SO2 oxidation reveal causes of seasonality in SO2 and sulfate isotopic composition, Environ. Sci. Technol., 47, 12174–12183, 2013b.
Hemingway, J., Olson, H., v Turchyn, A., Tipper, E., and Johnston, D. T.: Interpreting triple oxygen isotope signals from geologically preserved sulfate: Insights from modern Himalayan rivers, AGU Fall Meeting, San Francisco, 597703, 2019.
Herrmann, H.: Kinetics of aqueous phase reactions relevant for atmospheric chemistry, Chem. Rev., 103, 4691–4716, 2003.
Holt, B. and Kumar, R.: Oxygen isotope fractionation for understanding the sulphur cycle, Stable Isotopes: Natural and Anthropogenic Sulphur in the Environmentm, John Wiley & Sons, 27–41, 1991.
Holt, B. D., Cunningham, P. T., and Kumar, R.: Oxygen isotopy of atmospheric sulfates, Environ. Sci. Technol., 15, 804–808, 1981.
Janssen, C., Guenther, J., Krankowsky, D., and Mauersberger, K.: Relative formation rates of 50 O3 and 52 O3 in 16O–18O mixtures, J. Chem. Phys., 111, 7179–7182, 1999.
Jenkins, K. A. and Bao, H.: Multiple oxygen and sulfur isotope compositions of atmospheric sulfate in Baton Rouge, LA, USA, Atmos. Environ., 40, 4528–4537, 2006.
Kampschulte, A. and Strauss, H.: The sulfur isotopic evolution of Phanerozoic seawater based on the analysis of structurally substituted sulfate in carbonates, Chem. Geol., 204, 255–286, 2004.
Klemm, W. and Siedel, H.: Evaluation of the origin of sulphate compounds in building stone by sulphur isotope ratio, Geological Society, London, Special Publications, 205, 419–429, 2002.
Klimont, Z., Smith, S. J., and Cofala, J.: The last decade of global anthropogenic sulfur dioxide: 2000–2011 emissions, Environ. Res. Lett., 8, https://doi.org/10.1088/1748-9326/8/1/014003, 2013.
Kloppmann, W., Bromblet, P., Vallet, J., Vergès-Belmin, V., Rolland, O., Guerrot, C., and Gosselin, C.: Building materials as intrinsic sources of sulphate: a hidden face of salt weathering of historical monuments investigated through multi-isotope tracing (B, O, S), Sci. Total Environ., 409, 1658–1669, 2011.
Kloppmann, W., Rolland, O., Proust, E., and Montech, A.: Soluble salt sources in medieval porous limestone sculptures: A multi-isotope (N, O, S) approach, Sci. Total Environ., 470, 559–566, 2014.
Kramar, S., Mirtič, B., Knöller, K., and Rogan-Šmuc, N.: Weathering of the black limestone of historical monuments (Ljubljana, Slovenia): Oxygen and sulfur isotope composition of sulfate salts, Appl. Geochem., 26, 1632–1638, 2011.
Labidi, J., Cartigny, P., Hamelin, C., Moreira, M., and Dosso, L.: Sulfur isotope budget (32S, 33S, 34S and 36S) in Pacific–Antarctic ridge basalts: A record of mantle source heterogeneity and hydrothermal sulfide assimilation, Geochim. Cosmochim. Ac., 133, 47–67, 2014.
Lee, C.-W., Savarino, J., Cachier, H., and Thiemens, M.: Sulfur (32S, 33S, 34S, 36S) and oxygen (16O, 17O, 18O) isotopic ratios of primary sulfate produced from combustion processes, Tellus B, 54, 193–200, 2002.
Lee, C. C. W. and Thiemens, M. H.: The δ17O and δ18O measurements of atmospheric sulfate from a coastal and high alpine region: A mass-independent isotopic anomaly, J. Geophys. Res.-Atmos., 106, 17359–17373, 2001.
Le Gendre, E., Martin, E., Villemant, B., Cartigny, P., and Assayag, N.: A simple and reliable anion-exchange resin method for sulfate extraction and purification suitable for multiple O-and S-isotope measurements, Rapid Commun. Mass Spectrom., 31, 137–144, 2017.
Li, L., Chen, Z. M., Zhang, Y. H., Zhu, T., Li, J. L., and Ding, J.: Kinetics and mechanism of heterogeneous oxidation of sulfur dioxide by ozone on surface of calcium carbonate, Atmos. Chem. Phys., 6, 2453–2464, https://doi.org/10.5194/acp-6-2453-2006, 2006.
Li, X., Bao, H., Gan, Y., Zhou, A., and Liu, Y.: Multiple oxygen and sulfur isotope compositions of secondary atmospheric sulfate in a mega-city in central China, Atmos. Environ., 81, 591–599, 2013.
Lin, Y., Sim, M. S., and Ono, S.: Multiple-sulfur isotope effects during photolysis of carbonyl sulfide, Atmos. Chem. Phys., 11, 10283–10292, https://doi.org/10.5194/acp-11-10283-2011, 2011.
Lin, M., Kang, S., Shaheen, R., Li, C., Hsu, S.-C., and Thiemens, M. H.: Atmospheric sulfur isotopic anomalies recorded at Mt. Everest across the Anthropocene, P. Natl. Acad. Sci. USA, 115, 6964–6969, 2018a.
Lin, M., Zhang, X., Li, M., Xu, Y., Zhang, Z., Tao, J., Su, B., Liu, L., Shen, Y., and Thiemens, M. H.: Five-S-isotope evidence of two distinct mass-independent sulfur isotope effects and implications for the modern and Archean atmospheres, P. Natl. Acad. Sci. USA, 115, 8541–8546, 2018b.
Lloyd, R.: Oxygen isotope behavior in the sulfate-water system, J. Geophys. Res., 73, 6099–6110, 1968.
Longinelli, A. and Bartelloni, M.: Atmospheric pollution in Venice, Italy, as indicated by isotopic analyses, Water Air Soil Pollut., 10, 335–341, 1978.
Lyons, J. R.: Transfer of mass-independent fractionation in ozone to other oxygen-containing radicals in the atmosphere, Geophys. Res. Lett., 28, 3231–3234, 2001.
Markovic, S., Paytan, A., Li, H., and Wortmann, U. G.: A revised seawater sulfate oxygen isotope record for the last 4 Myr, Geochim. Cosmochim. Ac., 175, 239–251, 2016.
Martin, E.: Volcanic plume impact on the atmosphere and climate: O-and S-isotope insight into sulfate aerosol formation, Geosciences, 8, 198, https://doi.org/10.3390/geosciences8060198, 2018.
Martin, E., Bekki, S., Ninin, C., and Bindeman, I.: Volcanic sulfate aerosol formation in the troposphere, J. Geophys. Res.-Atmos., 119, 12660–12673, 2014.
Mauersberger, K., Erbacher, B., Krankowsky, D., Günther, J., and Nickel, R.: Ozone isotope enrichment: Isotopomer-specific rate coefficients, Science, 283, 370–372, 1999.
Montana, G., Randazzo, L., Oddo, I. A., and Valenza, M.: The growth of “black crusts” on calcareous building stones in Palermo (Sicily): a first appraisal of anthropogenic and natural sulphur sources, Environ. Geol., 56, 367–380, 2008.
Montana, G., Randazzo, L., and Mazzoleni, P.: Natural and anthropogenic sources of total suspended particulate and their contribution to the formation of black crusts on building stone materials of Catania (Sicily), Environ. Earth Sci., 67, 1097–1110, 2012.
Nielsen, H.: Isotopic composition of the major contributors to atmospheric sulfur, Tellus, 26, 213–221, 1974.
Oduro, H., Harms, B., Sintim, H. O., Kaufman, A. J., Cody, G., and Farquhar, J.: Evidence of magnetic isotope effects during thermochemical sulfate reduction, P. Natl. Acad. Sci. USA, 108, 17635–17638, 2011.
Ono, S., Eigenbrode, J. L., Pavlov, A. A., Kharecha, P., Rumble III, D., Kasting, J. F., and Freeman, K. H.: New insights into Archean sulfur cycle from mass-independent sulfur isotope records from the Hamersley Basin, Australia, Earth Planet. Sc. Lett., 213, 15–30, 2003.
Ono, S., Wing, B., Johnston, D., Farquhar, J., and Rumble, D.: Mass-dependent fractionation of quadruple stable sulfur isotope system as a new tracer of sulfur biogeochemical cycles, Geochim. Cosmochim. Ac., 70, 2238–2252, 2006a.
Ono, S., Wing, B., Rumble, D., and Farquhar, J.: High precision analysis of all four stable isotopes of sulfur (32S, 33S, 34S and 36S) at nanomole levels using a laser fluorination isotope-ratio-monitoring gas chromatography – mass spectrometry, Chem. Geol., 225, 30–39, 2006b.
Ono, S., Whitehill, A., and Lyons, J.: Contribution of isotopologue self-shielding to sulfur mass-independent fractionation during sulfur dioxide photolysis, J. Geophys. Res.-Atmos., 118, 2444–2454, 2013.
Patris, N., Delmas, R., Legrand, M., De Angelis, M., Ferron, F. A., Stiévenard, M., and Jouzel, J.: First sulfur isotope measurements in central Greenland ice cores along the preindustrial and industrial periods, J. Geophys. Res.-Atmos., 107, ACH 6-1–ACH 6-11, 2002.
Raab, M. and Spiro, B.: Sulfur isotopic variations during seawater evaporation with fractional crystallization, Chem. Geol., 86, 323–333, 1991.
Rees, C., Jenkins, W., and Monster, J.: The sulphur isotopic composition of ocean water sulphate, Geochim. Cosmochim. Ac., 42, 377–381, 1978.
Rennie, V. C. and Turchyn, A. V.: The preservation of δSSO434 and δOSO418 in carbonate-associated sulfate during marine diagenesis: A 25 Myr test case using marine sediments, Earth Planet. Sc. Lett., 395, 13–23, 2014.
Rodriguez-Navarro, C. and Sebastian, E.: Role of particulate matter from vehicle exhaust on porous building stones (limestone) sulfation, Sci. Total Environ., 187, 79–91, 1996.
Romero, A. B. and Thiemens, M. H.: Mass-independent sulfur isotopic compositions in present-day sulfate aerosols, J. Geophys. Res.-Atmos., 108, https://doi.org/10.1029/2003JD003660, 2003.
Sáiz-Jiménez, C.: Deposition of anthropogenic compounds on monuments and their effect on airborne microorganisms, Aerobiologia, 11, 161–175, 1995.
Savarino, J. and Thiemens, M. H.: Analytical procedure to determine both δ18O and δ17O of H2O2 in natural water and first measurements, Atmos. Environ., 33, 3683–3690, 1999.
Savarino, J., Lee, C. C., and Thiemens, M. H.: Laboratory oxygen isotopic study of sulfur (IV) oxidation: Origin of the mass-independent oxygen isotopic anomaly in atmospheric sulfates and sulfate mineral deposits on Earth, J. Geophys. Res.-Atmos., 105, 29079–29088, 2000.
Scheerer, S., Ortega-Morales, O., and Gaylarde, C.: Microbial Deterioration of Stone Monuments–An Updated Overview, Adv. Appl. Microbiol., 66, 97–139, 2009.
Schiavon, N.: Biodeterioration of calcareous and granitic building stones in urban environments, Geological Society, London, Special Publications, 205, 195–205, 2002.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: from air pollution to climate change, John Wiley & Sons, 2016.
Shaheen, R., Abaunza, M. M., Jackson, T. L., McCabe, J., Savarino, J., and Thiemens, M. H.: Large sulfur-isotope anomaly in nonvolcanic sulfate aerosol and its implications for the Archean atmosphere, P. Natl. Acad. Sci. USA, 111, 11979–11983, 2014.
Siegesmund, S., Török, A., Hüpers, A., Müller, C., and Klemm, W.: Mineralogical, geochemical and microfabric evidences of gypsum crusts: a case study from Budapest, Environ. Geol., 52, 385–397, 2007.
Sofen, E. D., Alexander, B., and Kunasek, S. A.: The impact of anthropogenic emissions on atmospheric sulfate production pathways, oxidants, and ice core , Atmos. Chem. Phys., 11, 3565–3578, https://doi.org/10.5194/acp-11-3565-2011, 2011.
Šrámek, J.: Determination of the source of surface deterioration on tombstones at the Old Jewish Cemetery in Prague, Stud. Conserv., 25, 47–52, 1980.
Stocker, T.: Climate change 2013: the physical science basis: Working Group I contribution to the Fifth assessment report of the Intergovernmental Panel on Climate Change, Cambridge University Press, 2014.
Thiemens, M. H.: Mass-independent isotope effects in planetary atmospheres and the early solar system, Science, 283, 341–345, 1999.
Thode, H., Monster, J., and Dunford, H.: Sulphur isotope geochemistry, Geochim. Cosmochim. Ac., 25, 159–174, 1961.
Thomazo, C., Brayard, A., Elmeknassi, S., Vennin, E., Olivier, N., Caravaca, G., Escarguel, G., Fara, E., Bylund, K., and Jenks, J.: Multiple sulfur isotope signals associated with the late Smithian event and the Smithian/Spathian boundary, Earth-Sci. Rev., 195, 96–113, https://doi.org/10.1016/j.earscirev.2018.06.019, 2018.
Thurston, R. S., Mandernack, K. W., and Shanks III, W. C.: Laboratory chalcopyrite oxidation by Acidithiobacillus ferrooxidans: oxygen and sulfur isotope fractionation, Chem. Geol., 269, 252–261, 2010.
Tiano, P.: Biodegradation of cultural heritage: decay mechanisms and control methods, in: Seminar article, New University of Lisbon, Department of Conservation and Restoration, 7–12, 2002.
Tiano, P., Bianchi, R., Gargani, G., and Vannucci, S.: Research on the presence of sulphurcycle bacteria in the stone of some historical buildings in Florence, Plant Soil, 43, 211–217, 1975.
Torfs, K. M., Van Grieken, R. E., and Buzek, F.: Use of stable isotope measurements to evaluate the origin of sulfur in gypsum layers on limestone buildings, Environ. Sci. Technol., 31, 2650–2655, 1997.
Turchyn, A. V., Schrag, D. P., Coccioni, R., and Montanari, A.: Stable isotope analysis of the Cretaceous sulfur cycle, Earth Planet. Sc. Lett., 285, 115–123, 2009.
Turro, N. J.: Influence of nuclear spin on chemical reactions: magnetic isotope and magnetic field effects (a review), P. Natl. Acad. Sci. USA, 80, 609–621, 1983.
Urey, H. C.: The thermodynamic properties of isotopic substances, J. Chem. Soc., 562–581, https://doi.org/10.1039/JR9470000562, 1947.
Vallet, J.-M., Gosselin, C., Bromblet, P., Rolland, O., Vergès-Belmin, V., and Kloppmann, W.: Origin of salts in stone monument degradation using sulphur and oxygen isotopes: First results of the Bourges cathedral (France), J. Geochem. Explor., 88, 358–362, 2006.
Vicars, W. C. and Savarino, J.: Quantitative constraints on the 17O-excess (Δ17O) signature of surface ozone: Ambient measurements from 50∘ N to 50∘ S using the nitrite-coated filter technique, Geochim. Cosmochim. Ac., 135, 270–287, 2014.
Weber, R., Chen, G., Davis, D., Mauldin III, R., Tanner, D., Eisele, F., Clarke, A., Thornton, D., and Bandy, A.: Measurements of enhanced H2SO4 and 3–4 nm particles near a frontal cloud during the First Aerosol Characterization Experiment (ACE 1), J. Geophys. Res.-Atmos., 106, 24107–24117, 2001.
Young, E. D., Galy, A., and Nagahara, H.: Kinetic and equilibrium mass-dependent isotope fractionation laws in nature and their geochemical and cosmochemical significance, Geochim. Cosmochim. Ac., 66, 1095–1104, 2002.