the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 09 Apr 2020
| 09 Apr 2020
Technical note: Preparation and purification of atmospherically relevant α-hydroxynitrate esters of monoterpenes
Elena Ali McKnight
Nicole P. Kretekos
Demi Owusu
Rebecca Lyn LaLonde
Organic nitrate esters are key products of terpene oxidation in the atmosphere. We report here the preparation and purification of nine nitrate esters derived from (+)-3-carene, limonene, α-pinene, β-pinene and perillic alcohol. The availability of these compounds will enable detailed investigations into the structure–reactivity relationships of aerosol formation and processing and will allow individual investigations into aqueous-phase reactions of organic nitrate esters.
- Article
(2457 KB) -
Supplement
(2684 KB) - BibTeX
- EndNote





Biogenic volatile organic compound (BVOC) emissions account for ∼88 % of non-methane VOC emissions. Of the total BVOC estimated by the Model of Emission of Gases and Aerosols from Nature version 2.1 (MEGAN2.1), isoprene is estimated to comprise half, and methanol, ethanol, acetaldehyde, acetone, α-pinene, β-pinene, limonene, ethene and propene together encompass another 30 %. Of the terpenoids, α-pinene alone is estimated to generate ∼66 Tg yr−1 (Guenther et al., 2012). These monoterpenes can be oxidized by nitrate radicals that are projected to account for more than half of the monoterpene-derived secondary organic aerosol (SOA) in the US (Pye et al., 2010). Nitrate oxidation pathways have been shown to be important particularly during nighttime. A large portion (30 %–40 %) of monoterpene emissions occur at night (Pye et al., 2010). These emissions can then react with NO3 radicals, formed from the oxidation of NO2 emissions by O3 (Pye et al., 2010).
The full role of organic nitrates (ONs) is complicated with many different sources and sinks (Perring et al., 2013). Fully deconvoluting the atmospheric processing of terpene-derived ON is difficult, particularly due to partitioning into the aerosol phase in which hydrolysis and other reactivity can occur (Bleier and Elrod, 2013; Rindelaub et al., 2014, 2015; Romonosky et al., 2015; Thomas et al., 2016). Hydrolysis reactions of nitrate esters of isoprene have been studied directly (Jacobs et al., 2014) and the hydrolysis of ON has been studied in bulk (Baker and Easty, 1950). These and other studies have shown that the hydrolysis of ON is dependent on structure (Darer et al., 2011). For example, primary and secondary ON are thought to be relatively stable (Hu et al., 2011). In contrast, tertiary nitrates have been shown to hydrolyze on the order of hours (Boyd et al., 2015; Liu et al., 2012) to minutes (Darer et al., 2011). To the best of our knowledge there is only one study of the hydrolysis of an isolated terpene-derived hydroxynitrate (2 in Fig. 1) (Rindelaub et al., 2016a).
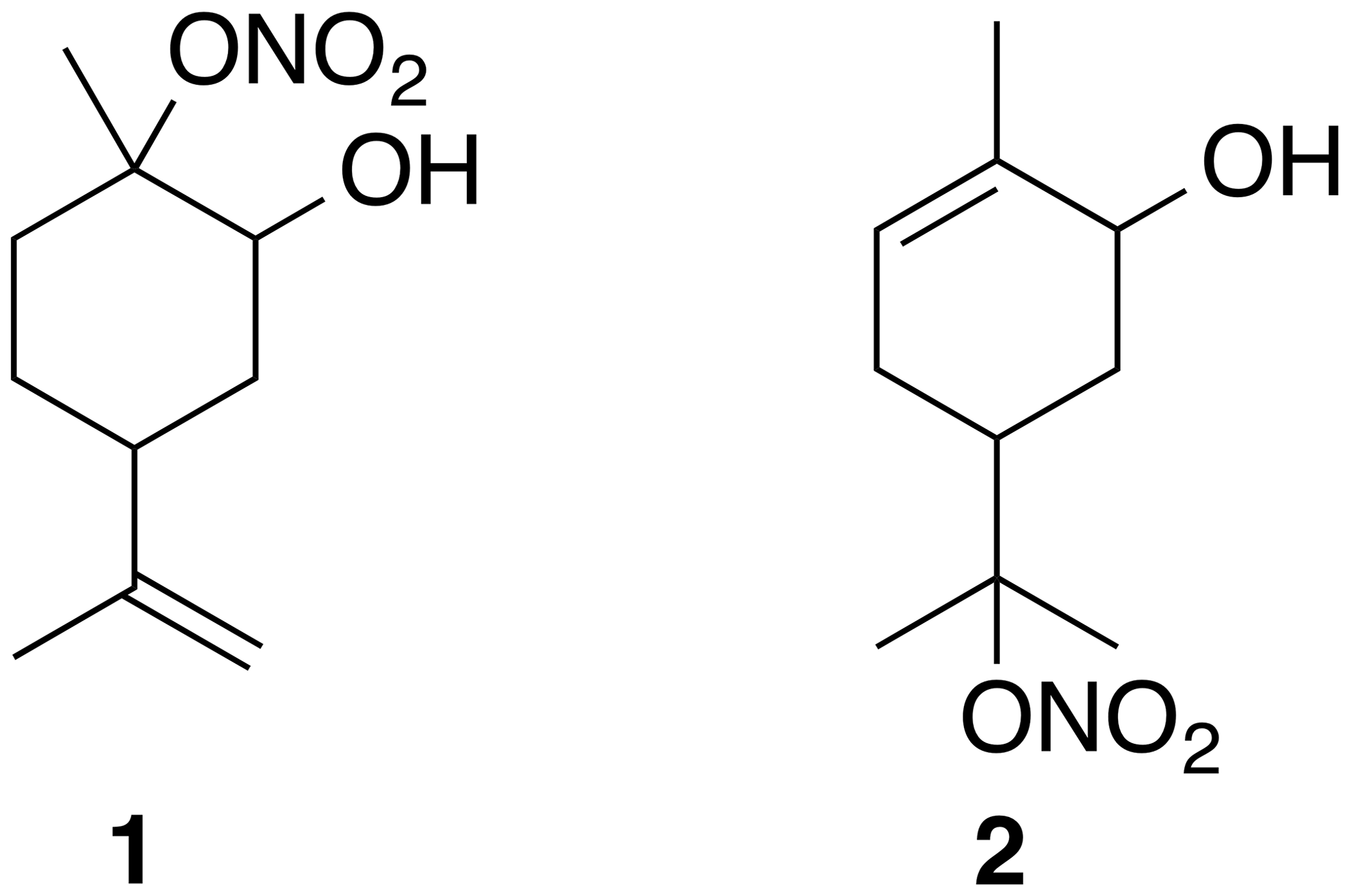
Figure 1Two hydroxynitrate esters with available spectral data. Relative stereochemistry is undefined.
Furthermore, fully understanding the atmospheric processes of organic molecules is restricted by the ability to identify these species (Nozière et al., 2015). Part of this challenge is, of course, related to the lack of available standards. While one certainly cannot synthesize all of the atmospherically relevant ON, having access to representative compounds from monoterpenes would enable key studies. With these molecules in hand, the atmospheric chemistry community could directly study the ON reactivity, such as hydrolysis, and deconvolute the structure–reactivity relationships. Additionally, novel method development would be enabled and validated (Rindelaub et al., 2016b). For example, Nozière and co-authors called attention to “the lack of NMR spectra libraries for atmospheric markers” as a barrier to utilizing nuclear magnetic resonance (NMR) spectroscopy in atmospheric science. While the preparation of α-hydroxynitrates of terpenes has been alluded to in a handful of reports, we have only been able to identify NMR data for two species (Fig. 1). Two main methods appear to have been attempted, treating an epoxide with either fuming nitric acid (Rollins et al., 2010) or bismuth nitrate (Rindelaub et al., 2016b; Romonosky et al., 2015). The utility of the former is limited by the extreme hazards involved with mixing fuming nitric acid with organic materials (Parker, 1995; Univ. of California, Berkeley, 2019) and only provides characterization data for a single compound. On the surface the latter appears to be a usable method but on closer inspection lacks spectral data, perhaps due to intractable and inseparable mixtures (Romonosky et al., 2015), and has been the subject of a retracted study (Pöschl et al., 2011).
2.1 Instrumentation and materials
All starting materials and reagents were purchased from commercial sources and used without further purification unless otherwise noted. Bismuth nitrate (98 %) was purchased from Strem Chemicals and ground to a fine powder prior to use. Isoprene, methyl vinyl ketone (MVK), (+)-3-carene, β-pinene, α-pinene oxide and 1,2-limonene oxide were purchased from Sigma-Aldrich. Silica gel chromatography was performed on a Teledyne Isco CombiFlash+ Lumen using 25 µm SiliCycle spherical silica gel. 1H and 13C NMR spectra were recorded on a Bruker AV400 spectrometer with chloroform (7.26 ppm) or benzene (7.28 ppm) as internal standards. Infrared (IR) spectra were recorded on a Thermo Nicolet IR100 spectrometer using a Thunderdome attenuated total reflectance (ATR) sample accessory. Melting points were collected on a DigiMelt 160 melting point apparatus. Exact masses were collected at the BioAnalytical Mass Spectrometry Facility at Portland State University using a ThermoElectron LTQ-Orbitrap Discovery high-resolution mass spectrometer with a dedicated Accela HPLC system. Low-resolution masses were collected on a Varian Saturn 2000 gas chromatographer–mass spectrometer (GC–MS) with a Restek Crossbind Rxi-5ms column. Abbreviations include meta-chloroperoxybenzoic acid (m-CPBA), hexanes (Hex), ethyl acetate (EtOAc), column volume (CV), thin layer chromatography (TLC), retention factor (Rf), diastereomeric ratio (d.r.), broad (br), singlet (s), doublet (d), doublet of doublets (dd), doublet of doublets of doublets (ddd), doublet of quartet (dq), triplet (t), multiplet (m), broad (b), weak (w), medium (m), strong (s) and very strong (vs).
2.2 Preparation of epoxides
2.2.1 trans-Carene oxide (3)
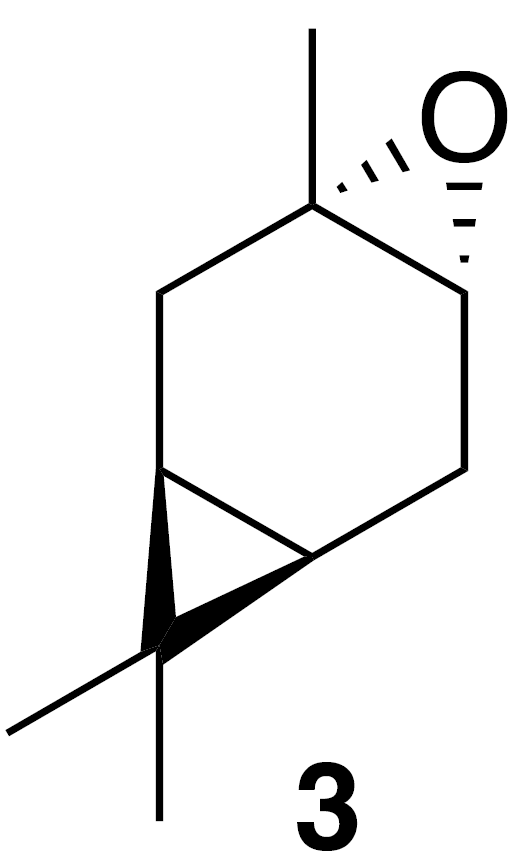
To a solution of (+)-3-carene (0.86 g, 1 mL, 7.34 mmol) in dichloromethane (25 mL) at 0 ∘C was added meta-chloroperoxybenzoic acid (m-CPBA) (1.85 g, 8.1 mmol, 1.1 equiv.). The solution was warmed to 23 ∘C over 1 h. The solution was poured onto saturated aqueous (sat. aq) sodium bicarbonate (30 mL) and extracted with dichloromethane (3×30 mL). The combined organics were washed with sat. aq sodium bicarbonate (2×30 mL), dried (MgSO4) and concentrated to yield a crude clear, colorless oil (1.17 g). The crude oil was purified by column chromatography (SiO2; 0 %–25 % EtOAc/Hex over 10 column volumes) to yield clear colorless oil (685 mg, 4.5 mmol; 61 % yield): 1H NMR (400 MHz, chloroform-d) δ 2.82 (s, 1 H), 2.28 (ddd, J=16.4, 9.0, 1.9 Hz, 1 H), 2.13 (dd, J=16.2, 9.1 Hz, 1 H), 1.63 (dt, J=16.4, 2.3 Hz, 1 H), 1.48 (dd, J=16.1, 2.3 Hz, 1 H), 1.24 (s, 3 H), 1.00 (s, 3 H), 0.72 (s, 3 H), 0.52 (td, J=9.1, 2.3 Hz, 1 H), 0.44 (td, J=9.1, 2.3 Hz, 1 H) ppm. Spectral data are consistent with literature reports (Cabaj et al., 2009).
2.2.2 cis-Carene oxide (6)
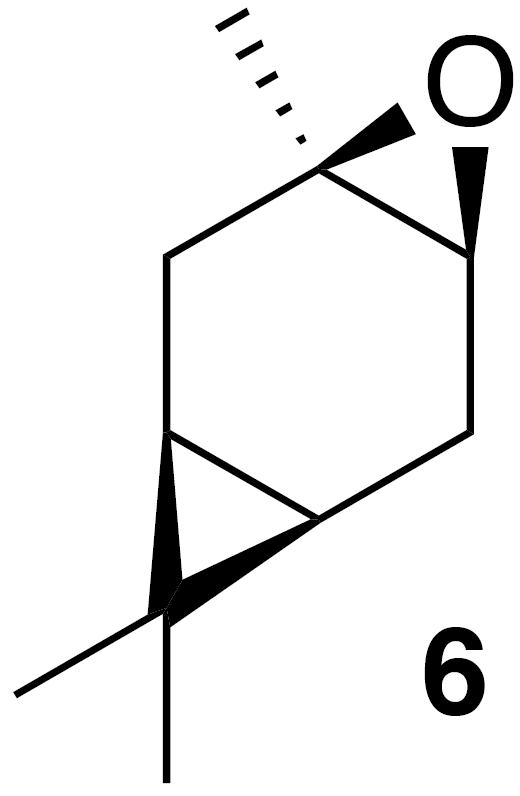
According to the methods of Cocker and Grayson (1969), a round bottom flask was charged with a solution of (+)-3-carene (2.7 g, 20 mmol) in dioxane (20 mL) and water (10 mL). Calcium carbonate (2 g, 20 mmol) and N-bromosuccinimide (7 g, 40 mmol) were added to the solution. The internal temperature rose to 50 ∘C after the initial addition. The mixture was stirred for 2 h then poured onto water (50 mL), filtered and washed with diethyl ether. The filtrate was extracted with ether (2×100 mL). The combined extract was washed with water (3×100 mL) and sodium thiosulfate (5 % aq, 20 mL), dried (Na2SO4), and concentrated to yield a crude pale yellow oil. Purification by column chromatography (SiO2; 0 %–50 % EtOAc ∕ Hex) yielded the bromohydrin as a white crystalline solid (4.6 g, 99 % yield): 1H NMR (400 MHz, chloroform-d) δ 4.07 (dd, J=11.1, 7.6 Hz, 1 H), 2.50–2.36 (m, 2 H), 2.21 (dd, J=14.6, 10.1 Hz, 1 H), 1.41 (dd, J=4.9, 1.2 Hz, 1 H), 1.38 (s, 3 H), 1.03 (s, 3 H), 1.00 (s, 3 H), 0.91–0.80 (m, 1 H), 0.70 (m, 1 H). Spectral data are consistent with literature reports (Cocker and Grayson, 1969). Bromohydrin (2.44 g, 10.5 mmol) was dissolved in 100 mL tBuOH (warmed with a water bath). Potassium tert-butoxide (2.44 g, 21.8 mmol, 2.08 equiv.) was added, and the solution was stirred at rt for 2 h. The solution was poured onto water (50 mL), extracted with diethyl ether (3×75 mL), washed with water (3×50 mL), dried (MgSO4) and concentrated to yield 2.5 g crude clear oil. Purification by column chromatography (40 g SiO2; 0 %–25 % EtOAc ∕ Hex over 10 CVs) yielded a clear colorless oil (1.31 g, 63 % yield): 1H NMR (400 MHz, chloroform-d) δ 2.89 (d, J=5.6 Hz, 1 H), 2.30 (ddd, J=16.7, 9.0, 5.6 Hz, 1 H), 2.08 (dd, J=16.4, 8.9 Hz, 1 H), 1.82 (s, 1 H), 1.78 (s, 1 H), 1.32 (s, 3 H), 0.98 (s, 3 H), 0.94 (s, 3 H), 0.63–0.49 (m, 2 H). Spectral data are consistent with literature reports (Cocker and Grayson, 1969).
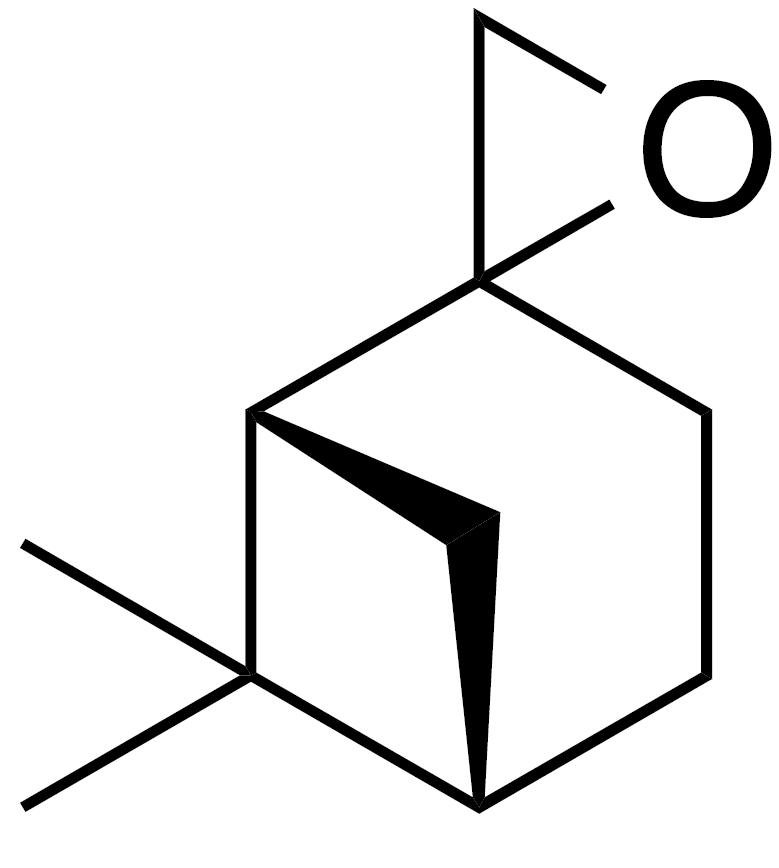
2.2.3 8,9-Limonene oxide (9)
![]()
Methyl vinyl ketone (10 mL, 90 mmol), isoprene (10 mL, 75 mmol) and dichloromethane (DCM) (90 mL) were added to a round bottom flask with stir bar. The flask was purged with inert atmosphere and chilled to 0 ∘C with stirring. AlCl3 (1.2 g, 9 mmol) was added in three portions over 10 min. The ice bath was removed and the reaction mixture was stirred for 1.5 h. The crude reaction mixture was filtered through a 1.5 in. (3.8 cm) silica gel pad (350 mL, 9 cm diameter) and washed with 8 % EtOAc : Hex (4×160 mL). The filtrate was concentrated to yield 1-(4-methyl-3-cyclohexene) ethenone (8 in Fig. 4) as a clear yellow liquid (9.68 g, 88 % yield): 1H NMR (400 MHz, chloroform-d) δ 5.44–5.38 (m, 1 H), 2.59–2.49 (m, 1 H), 2.15–2.20 (s, 4 H), 2.06–1.95 (m, 3 H), 1.67 (s, 3 H), 1.61 (m, 2 H). Spectral data are consistent with literature reports (Buss et al., 1987). Sodium hydride (1.766 g, 44.16 mmol) was suspended in dry dimethylsulfoxide (DMSO) (25 mL). A solution of trimethylsulfonium iodide (10.356 g, 47.06 mmol) in DMSO (50 mL) was added via cannula. The solution was stirred at rt for 30 min and then heated to 50 ∘C until gas evolution ceased (1 h). To this solution was added 1-(4-methyl-3-cyclohexen-1-yl) ethenone (8 in Fig. 4) (5 g, 36 mmol) and heated to 70 ∘C for 2 h. The reaction mixture was cooled to room temperature and poured onto sat. aq. NH4Cl (100 mL), extracted with methyl tert-butyl ether (MTBE) (4×100 mL). The combined organics were washed with water (4×50 mL), dried (MgSO4) and concentrated to yield 5.1 g crude oil. Purification by column chromatography (80 g SiO2; 0 %–20 % EtOAc ∕ Hex over 10 CVs) yielded 8,9-limonene oxide (1 : 1 d.r.) as a clear, colorless oil (3.76 g, 68 % yield): 1H NMR (400 MHz, chloroform-d) δ 5.43–5.33 (br s, 1 H), 2.67 (t, J=5.3 Hz, 1 H), 2.58 (dd, J=10.9, 4.8 Hz, 1 H), 2.17–1.73 (m, 5 H), 1.66 (s, 3 H), 1.44 (m, 2 H), 1.29 (d, J=4.5 Hz, 3 H). Spectral data are consistent with literature reports (Almeida and Jones Jr., 2005)
2.2.4 cis-1,2-Limonene oxide (12-cis)
![]()
According to the methods of Steiner et al. (2002), a 20 mL scintillation vial was charged with 1,2-limonene oxide, pyrrolidine and water. The vial was sealed with a Teflon-lined cap and heated to 88 ∘C for 24 h. The vial was cooled to rt, transferred to a separatory funnel with 100 mL pentane, and washed with sat. aq. ammonium chloride (3×30 mL) and water (1×50 mL). The organics were dried (MgSO4) and concentrated to yield a light yellow oil. Purification by column chromatography (80 g spherical SiO2; 0 %–15 % EtOAc ∕ Hex over 10 CVs) yielded cis-1,2-limonene oxide as a clear, colorless oil. (1.52 g, 64 %): 1H NMR (400 MHz, chloroform-d) δ 4.77–4.71 (m, 1 H), 4.69 (s, 1 H), 3.07 (s, 1 H), 2.22–2.07 (m, 2 H), 1.90–1.84 (m, 2 H), 1.75–1.64 (m, 5 H), 1.33 (s, 3 H), 1.28–1.15 (m, 1 H) ppm. Spectral data are consistent with literature reports (Steiner et al., 2002).
2.2.5 trans-1,2-Limonene oxide (12-trans)
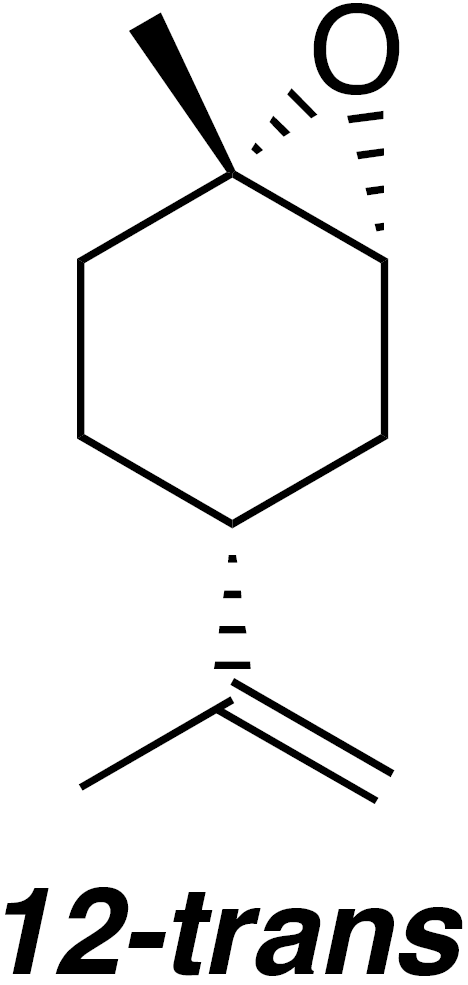
According to the methods of Steiner et al. (2002), a 50 mL flask was charged with 1,2-limonene oxide (4.57 g, 30 mmol), pyrazole (0.34 g, 5 mmol) and water (16.2 mL). The reaction was heated to reflux for 5 h. The reaction mixture was cooled to 80 ∘C and transferred to a separatory funnel. Warm water (80 ∘C; 60 mL) was added. The emulsion was extracted with pentane (3×50 mL). The combined organic layers were dried (MgSO4) and concentrated to yield 1.481 g crude oil. Purification by column chromatography (80 g spherical SiO2; 0 %–20 % EtOAc ∕ Hex over 13.5 CVs) yielded trans-1,2-limonene oxide (485 mg, 20 % yield): 1H NMR (400 MHz, chloroform-d) δ 4.68 (s, 2 H), 3.01 (d, J=5.3 Hz, 1 H), 2.11–2.00 (m, 2 H), 1.96–1.82 (m, 1 H), 1.75–1.64 (m, 5 H), 1.45–1.36 (m, 2 H), 1.34 (s, 3 H) ppm. Spectral data are consistent with literature reports (Steiner et al., 2002).
2.2.6 Perillic alcohol epoxide (15)
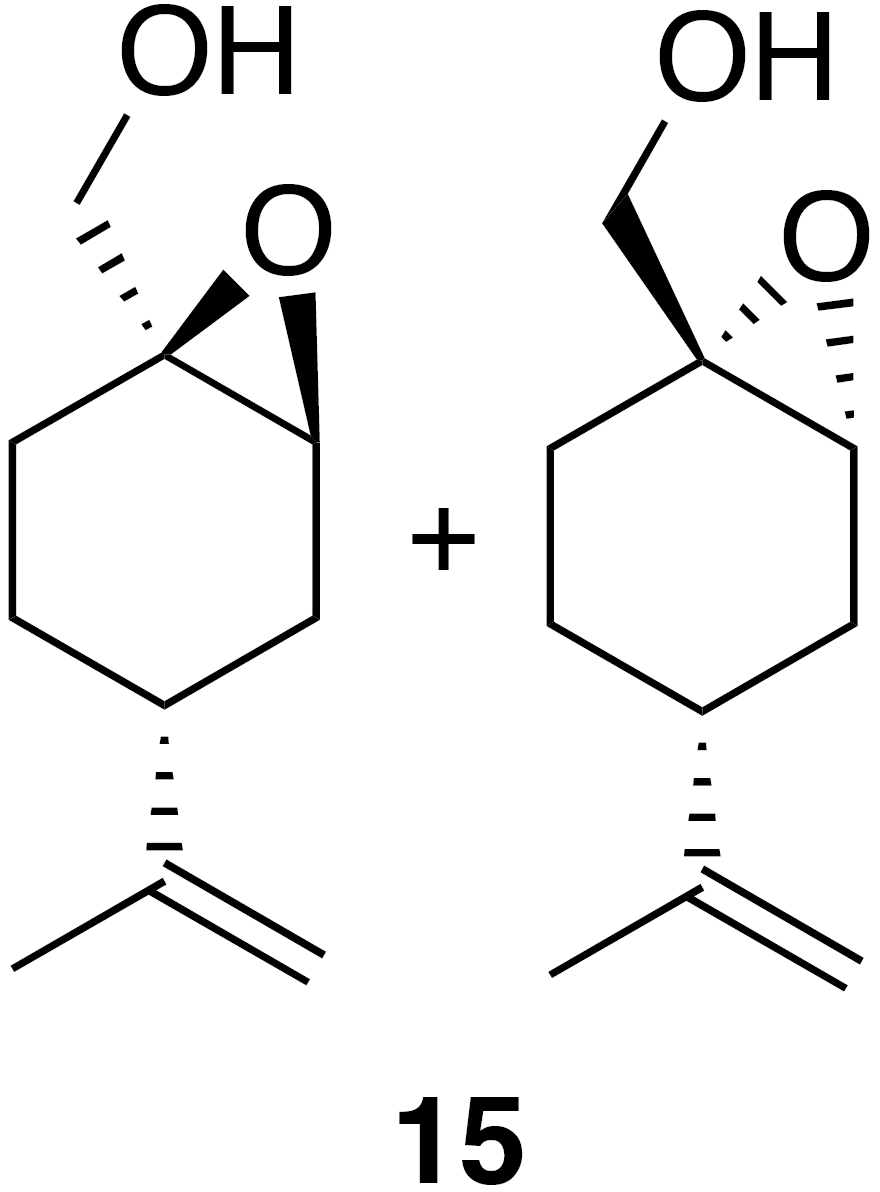
Perillic alcohol (5 mL, 31.46 mmol) was combined with dichloromethane (62 mL) in a round bottom flask with stir bar and cooled to 0 ∘C under inert atmosphere. m-CPBA (5.97 g, 34.61 mmol) was added in five portions over the course of 10 min. After 30 min the reaction was warmed back up to room temperature and allowed to stir for an hour at room temperature. The reaction mixture was filtered through celite, washed with DCM (3×30 mL) and then transferred to a separatory funnel where it was washed with sodium bicarbonate (50 mL) and brine (50 mL). The organic layer was dried (Na2SO4) and concentrated to yield a crude oil (5.1 g). Purification by column chromatography (80 g spherical SiO2; 0 %–100 % EtOAc ∕ Hex over 15 CVs) yielded the epoxide (15) as a clear, colorless oil (3.363 g, 19.8 % yield): 1H NMR (400 MHz, chloroform-d) δ 4.77–4.66 (m, 2 H), 3.78–3.59 (m, 2 H), 3.41–3.31 (m, 1 H), 2.26–2.14 (m, 1 H), 2.09 (doublet of triplet of doublet (dtd) J=14.9, 5.6, 1.8 Hz, 1 H), 1.88–1.83 (m, 1 H), 1.80–1.59 (m, 7 H), 1.56–1.38 (m, 1 H), 1.32–1.16 (m, 1 H). Spectral data are consistent with literature reports (Thomas et al., 2016).
2.2.7 β-Pinene oxide
Potassium peroxide monosulfate (4.750 g, 31.20 mmol) was dissolved in deionized water (60 mL). Sodium bicarbonate (4.019 g, 47.84 mmol) was placed into a 125 mL Erlenmeyer flask and acetone (40 mL) was added, followed by β-pinene (1.59 mL, 9.974 mmol). The solution of potassium peroxide monosulfate mixture was slowly added into the β-pinene mixture over the course of 3 min via syringe while the mixture was stirring at 800 rpm. The reaction mixture was stirred for exactly 30 min while stirring at 800 rpm at room temperature. The reaction mixture was transferred to a separatory funnel and extracted with dichloromethane (2×40 mL), dried (MgSO4) and concentrated to produce a clear oil (1500 g, 96 % yield). 1H NMR (400 MHz, chloroform-d) δ 2.79 (d, J=4.8 Hz, 1 H), 2.62 (d, J=4.8 Hz, 1 H), 2.30 (dtd, J=10.3, 5.9, 1.5 Hz, 1 H), 2.19 (ddd, J=15.1, 10.7, 8.1 Hz, 1 H), 2.06–1.97 (m, 1 H), 1.97–1.79 (m, 2 H), 1.75–1.65 (m, 2 H), 1.53 (t, J=5.4 Hz, 1 H), 1.27 (s, 3 H), 0.94 (s, 3 H). Spectral data are consistent with literature reports (Charbonneau et al., 2018).
2.3 General nitration method
A round bottom flask was charged with a solution of epoxide (2 mmol) in toluene, dioxane or dichloromethane (10 mL, 0.2 M). Bismuth nitrate (1.164 g, 2.4 mmol, 1.2 equiv.) was added to the reaction mixture. The reaction mixture was stirred for 30–60 min. When TLC indicated complete consumption of starting material, the reaction was filtered through a 1 in. (2.5 cm) celite pad and washed with DCM (2×15 mL). The filtrate was transferred to a separatory funnel and washed with sodium bicarbonate (3×15 mL). The organic layer was dried with sodium sulfate, filtered and concentrated to yield a crude liquid. Desired products were isolated via column chromatography.
2.3.1 Nitrate ester (4)
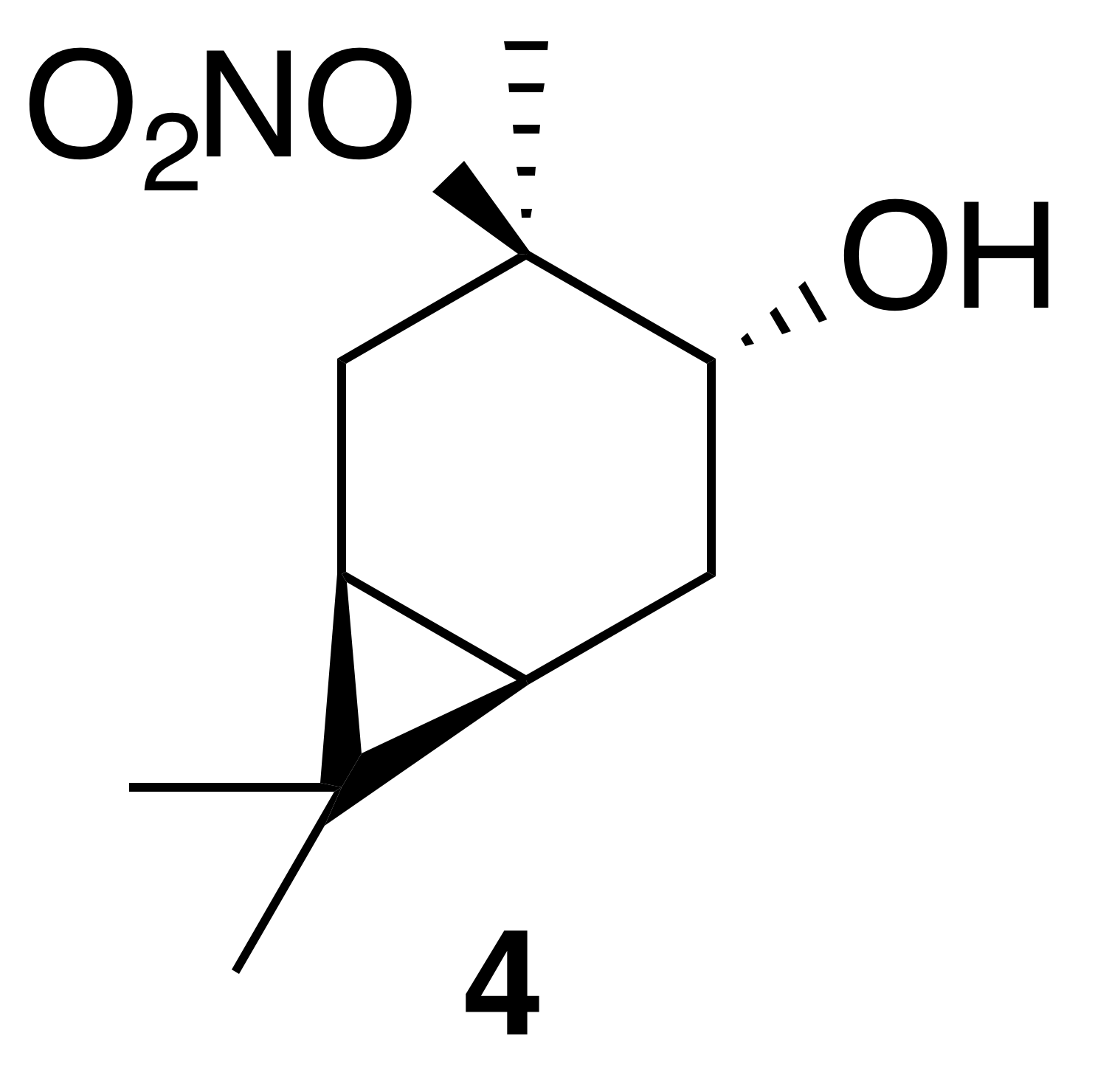
Nitration of trans-3-carene oxide (3 mmol) was carried out according to the general method with dioxane. Purification of the crude oil by column chromatography (12 g SiO2, 0 %–30 % EtOAc ∕ Hex) yielded nitrate ester (4) as a white crystalline solid (345 mg, 53 % yield): mp = 52.2–54.0 ∘C; IR (ATR) cm−1 3412 (br w, OH), 2944 (w), 2360 (w), 1616 (vs, NO2), 1291 (m, NO2), 868 (m, NO2); high-resolution mass spectrometry (HRMS) electrospray ionization (ESI) + m∕z calculated for [C10H18O4N]+ (216.1230, observed; 216.1236, expected) 1H NMR (400 MHz, chloroform-d) δ 3.70 (dd, J=9.6, 7.6 Hz, 1 H), 2.86–2.78 (m, 1 H), 2.16 (dd, J=14.8, 7.5 Hz, 1 H), 1.83 (ddd, J=14.8, 9.6, 7.9 Hz, 1 H), 1.65 (s, 3 H), 1.47 (dd, J=14.3, 4.1 Hz, 1 H), 1.03 (s, 3 H), 1.01 (s, 3 H), 0.83–0.73 (m, 2 H); 13C NMR (101 MHz, chloroform-d) δ 95.68, 71.03, 28.37, 28.34, 27.02, 19.95, 18.98, 17.95, 15.70, 14.39 ppm.
2.3.2 cis-4-Caranone (7)
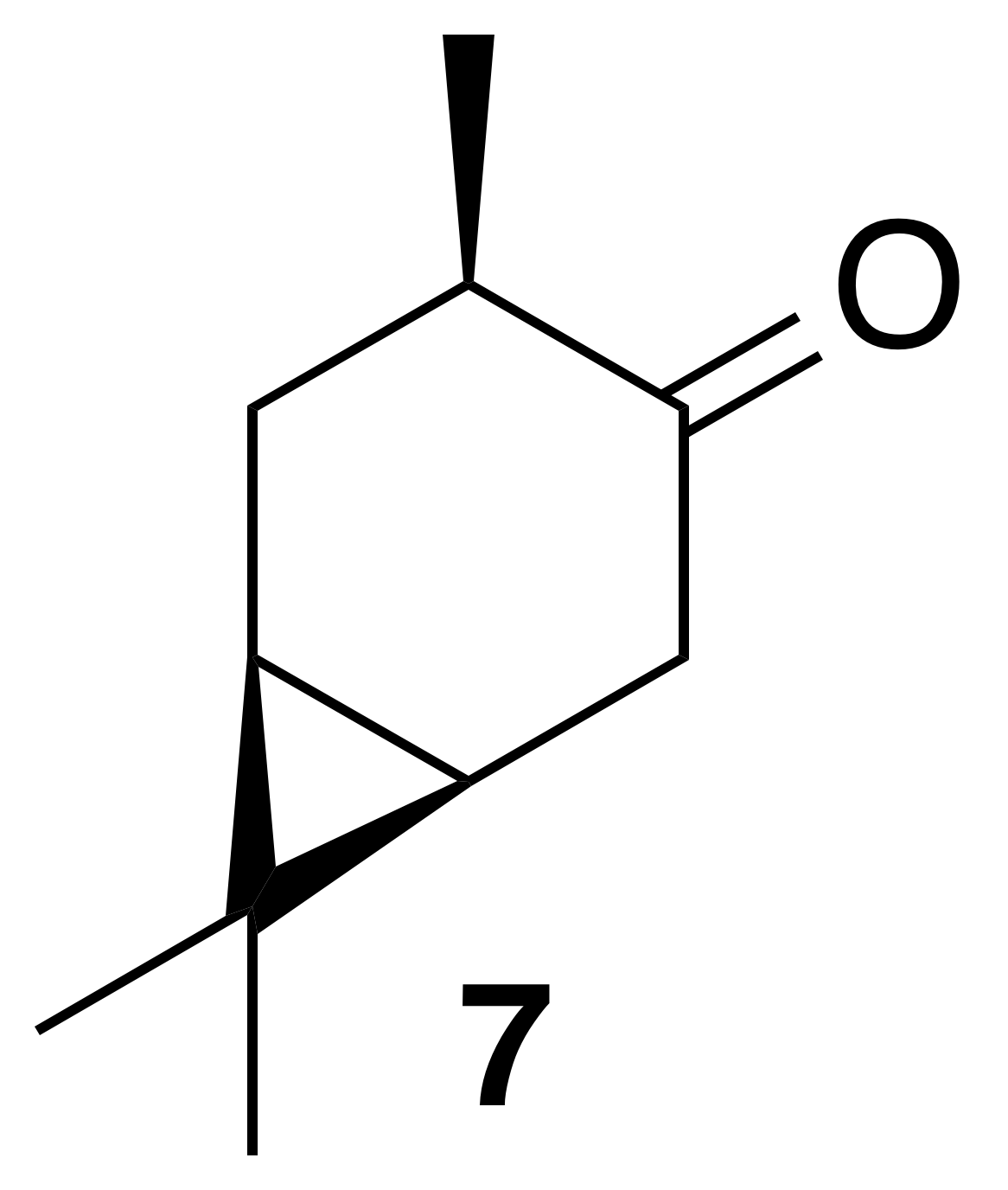
Nitration of cis-3-carene oxide was attempted according to the general method with dioxane or dichloromethane. Purification of the crude oil by column chromatography (12 g SiO2, 0 %–40 % EtOAc ∕ Hex) yielded cis-4-caranone as a clear, colorless oil (53 % yield): Rf 0.51 (5 % EtOAc : Hex; anisaldehyde); 1H NMR (400 MHz, chloroform-d) δ 2.54 (ddd, J=18.0, 8.4, 1.0 Hz, 1 H), 2.44–2.25 (m, 3 H), 1.33–1.20 (m, 1 H), 1.14–0.99 (m, 5 H), 0.97 (d, J=6.4 Hz, 3 H), 0.86 (s, 3 H); 13C NMR (101 MHz, chloroform-d) δ 216.79, 41.99, 36.84, 29.77, 27.91, 22.83, 20.34, 19.47, 14.89, 14.11 ppm. Spectral data are consistent with literature reports (Kolehmainen et al., 1993).
2.3.3 Reaction of 8,9-limonene oxide (9)
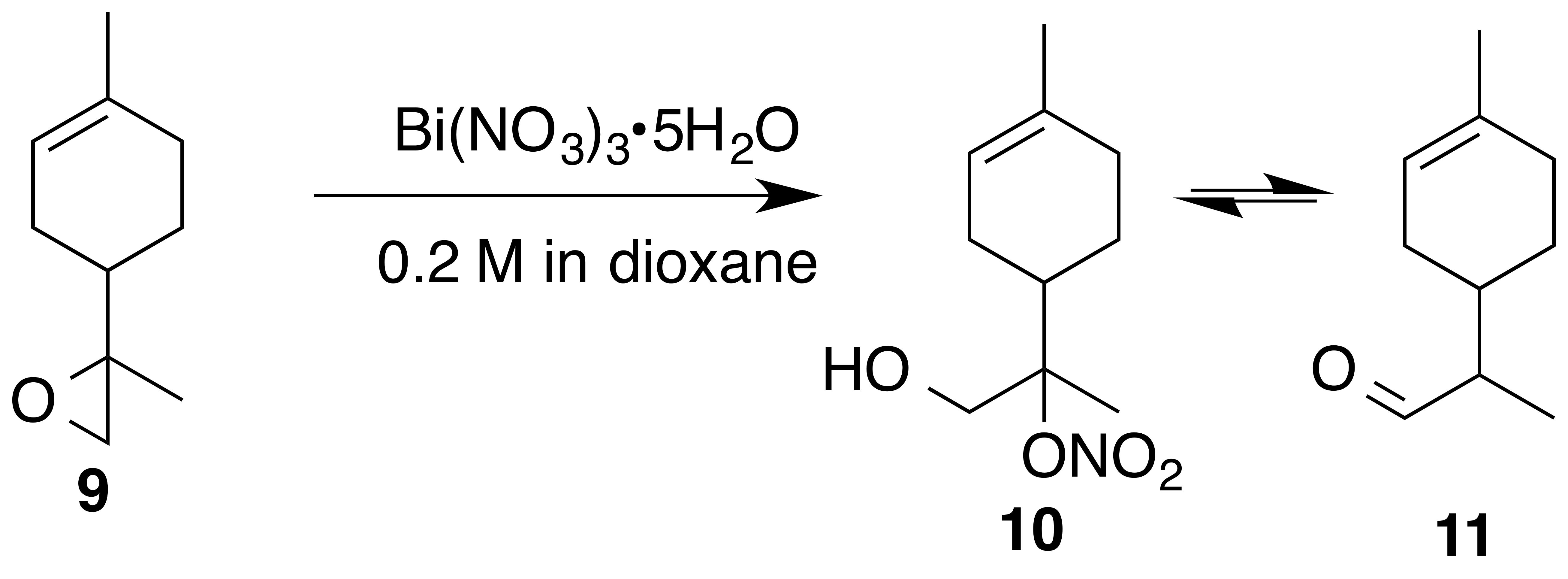
Nitration of 8,9-limonene oxide (9) was attempted according to the general method with acetonitrile or dioxane. Dioxane yielded a mixture of putative nitrate ester (10) and aldehyde (11) (1 : 0.25) as a crude oil. Nitrate (10) (1 : 1 d.r.): Rf 0.25 (15 % EtOAc : Hex; anisaldehyde); IR (ATR) cm−1 3417 (br), 2913 (w), 1613 (vs, NO2), 1439 (w), 1289 (m, NO2), 865 (s, NO2); 1H NMR (400 MHz, chloroform-d) δ 5.39–5.35 (m, 1 H), 4.06–3.86 (m, 2 H), 2.41–2.26 (m, 1 H), 2.09–1.82 (m, 5 H), 1.66 (s, 3 H), 1.49 (s, 1.5 H), 1.48 (s, 1.5 H), 1.43–1.31 (m, 2 H) ppm; 13C NMR (101 MHz, chloroform-d) δ 134.31, 134.01, 119.57, 119.31, 97.40, 97.17, 64.62, 64.27, 38.24, 38.19, 30.58, 30.47, 26.42, 25.87, 24.19, 23.32, 23.25, 23.22, 17.17, 16.78 ppm. Purification by column chromatography (12 g SiO2 gold column, 0 %–50 % EtOAc ∕ Hex over 30 CVs) yielded acetaldehyde (11) (1 : 1 d.r) as a clear, colorless oil (105 mg, 35 % yield): 1H NMR (400 MHz, chloroform-d) δ 9.69 (d, J=2.4 Hz, 0.5 H), 9.68 (d, J=2.7 Hz, 0.5 H), 5.42–5.35 (m, 1 H), 2.35–2.25 (m, 1 H), 2.10–1.73 (m, 6 H), 1.68–1.62 (br s, 3 H), 1.46–1.32 (m, 1 H), 1.10 (s, 1.5 H), 1.08 (s, 1.5 H) ppm; 13C NMR (101 MHz, chloroform-d) δ 205.55, 205.47, 134.09, 134.05, 119.95, 119.88, 51.00, 50.68, 34.32, 34.25, 30.15, 29.99, 29.67, 28.05, 27.31, 25.43, 23.43, 10.34, 10.24 ppm. Spectral data are consistent with literature reports (Uehara et al., 2017; Reid and Watson, 2018).
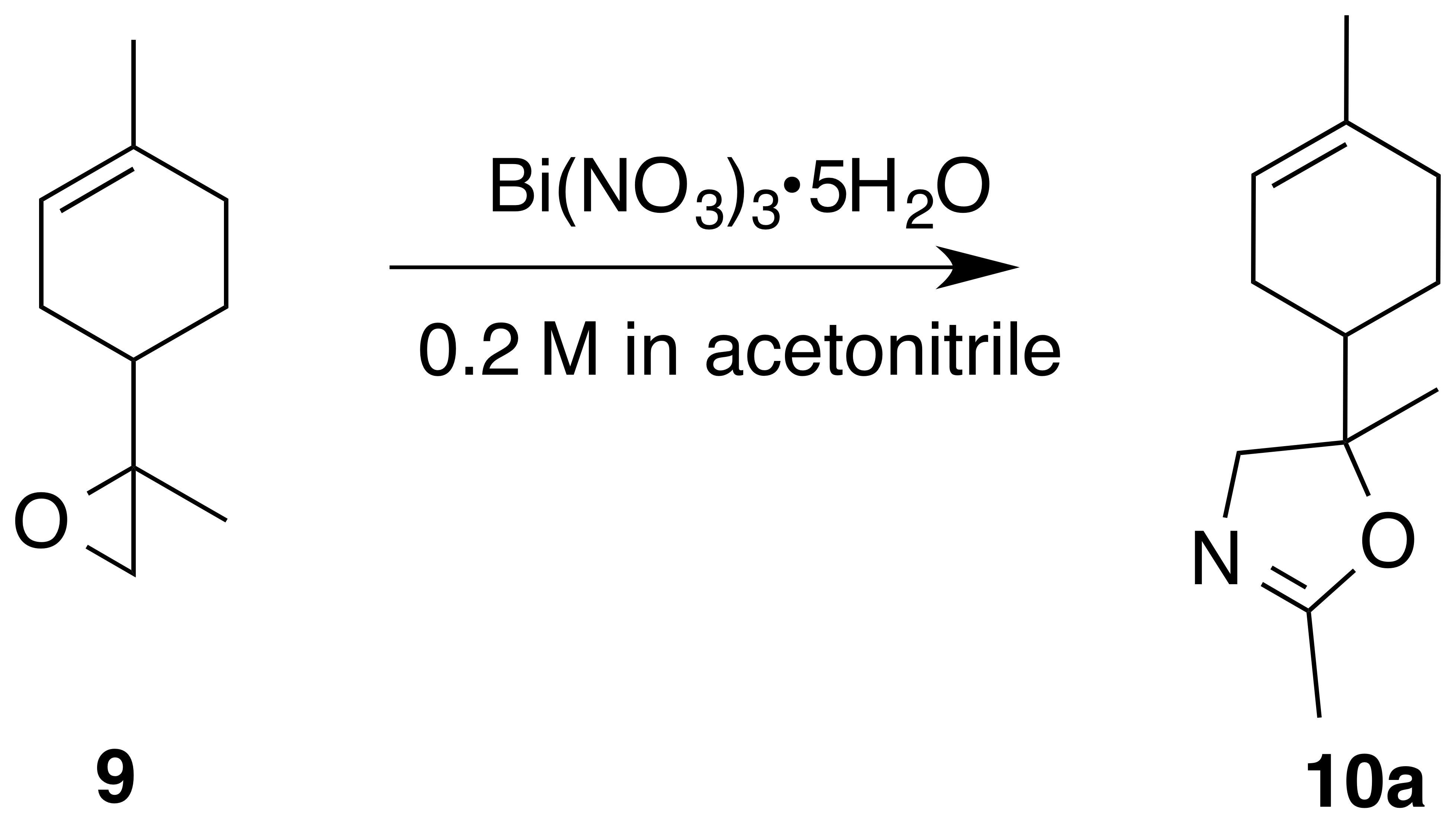
Acetonitrile yielded oxazoline (10a) (1 : 1 d.r.) as a clear, colorless oil (76 % yield): Rf 0.46 (75 % EtOAc : Hex; anisaldehyde); IR (ATR) cm−1 3323 (br), 2963 (m), 2913 (m), 1674 (vs), 1437 (m), 1384 (m), 1265 (m), 991 (vs), 915 (m); 1H NMR (400 MHz, chloroform-d) δ 5.38 (s, 1 H), 4.10 (d, J=6.2 Hz, 0.5 H), 4.08 (d, J=6.1 Hz, 0.5 H), 3.82 (d, J=8.3 Hz, 0.5 H), 3.79 (d, J=8.3 Hz, 0.5 H), 2.16–1.91 (m, 6 H), 1.89–1.75 (m, 1 H), 1.73–1.56 (m, 5 H), 1.29–1.14 (m, 4 H); 13C NMR (101 MHz, chloroform-d) 162.73, 162.64, 134.23, 133.89, 120.28, 120.23, 77.22, 76.08, 75.39, 72.75, 72.60, 42.55, 42.28, 30.65, 30.57, 26.75, 26.39, 25.03, 24.58, 23.79, 23.75, 23.37, 13.88, 13.87 ppm; GC–MS (100–275 ∘C; 15 ∘C min−1): 6.691 min, m∕z 193.8 (M+, 100 %), 135.0 (M − [C2H3NO], 10 %).
2.3.4 Nitrate ester (13)
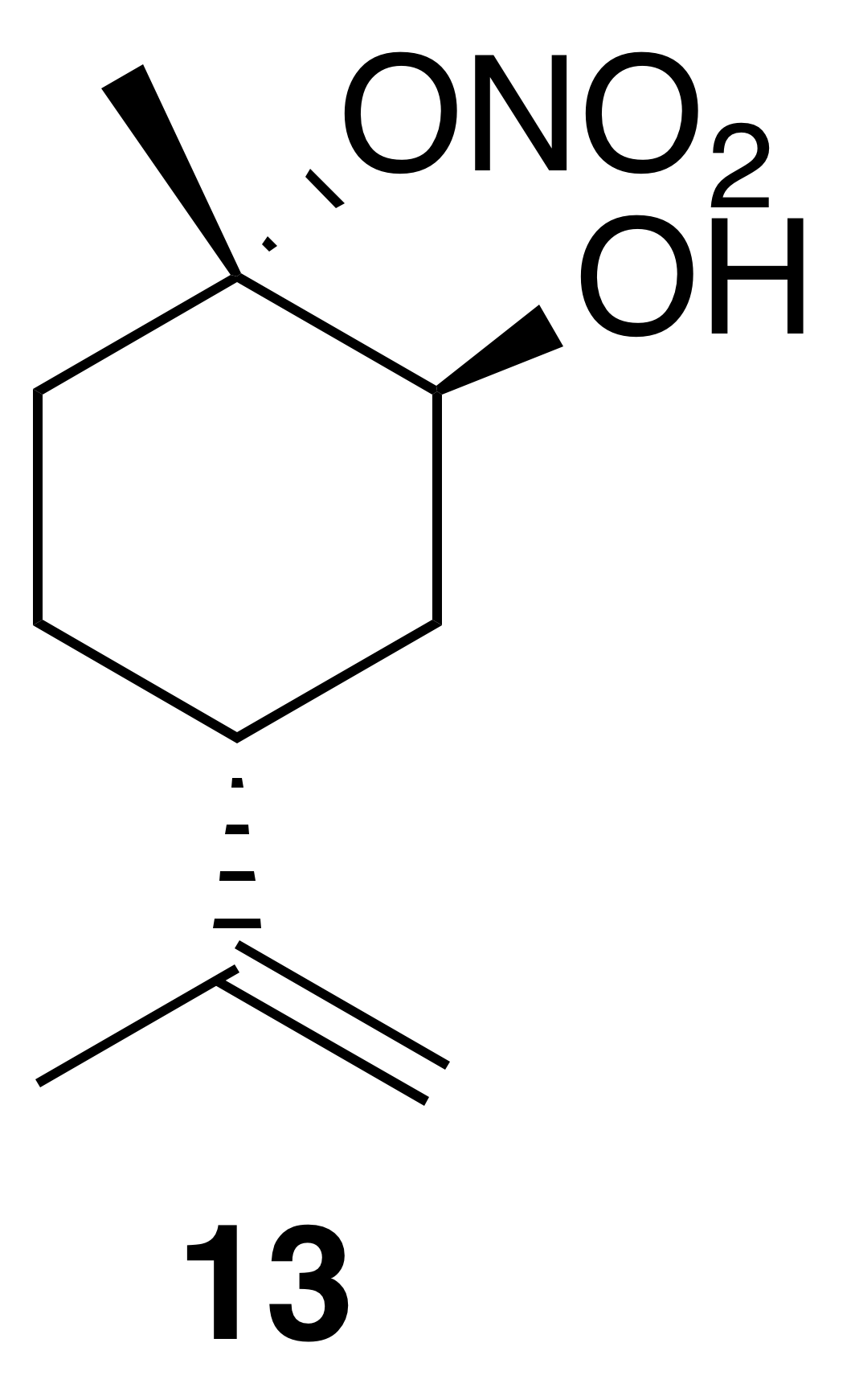
Nitration of cis-1,2-limonene oxide was carried out according to the general method with dioxane or dichloromethane. Purification by column chromatography (12 g SiO2 gold column, 0 %–50 % EtOAc ∕ Hex over 30 CVs) yielded nitrate ester (13) as a clear, colorless oil (53 % yield in dioxane; 62 % yield in DCM): IR (ATR) cm−1 3660 (w, alcoholic OH), 3340 (bd w, alcoholic OH), 2980 (m), 1618 (s, NO2), 1291 (m, NO2), 860 (vs, NO2); HRMS ESI + m∕z calculated for [C10H17O4NNa]+ (238.1052, observed; 238.1055, expected); 1H NMR (400 MHz, chloroform-d) δ 4.76 (dd, J=8.1, 1.5 Hz, 2 H), 4.13 (s, 1 H), 2.35 (doublet of doublet of triplet (ddt), J=11.6, 8.4, 4.1 Hz, 1 H), 2.24 (dtd, J=14.8, 3.7, 1.4 Hz, 1 H), 1.98–1.78 (m, 3 H), 1.76–1.73 (m, 4 H), 1.68–1.58 (m, 4 H), 1.49 (triplet of doublet of doublet (tdd), J=13.3, 11.6, 3.6 Hz, 1 H); 13C NMR (101 MHz, chloroform-d) δ 148.49, 109.48, 91.34, 69.22, 36.84, 33.86, 29.90, 25.77, 20.98, 20.93 ppm.
2.3.5 Nitrate ester (14)
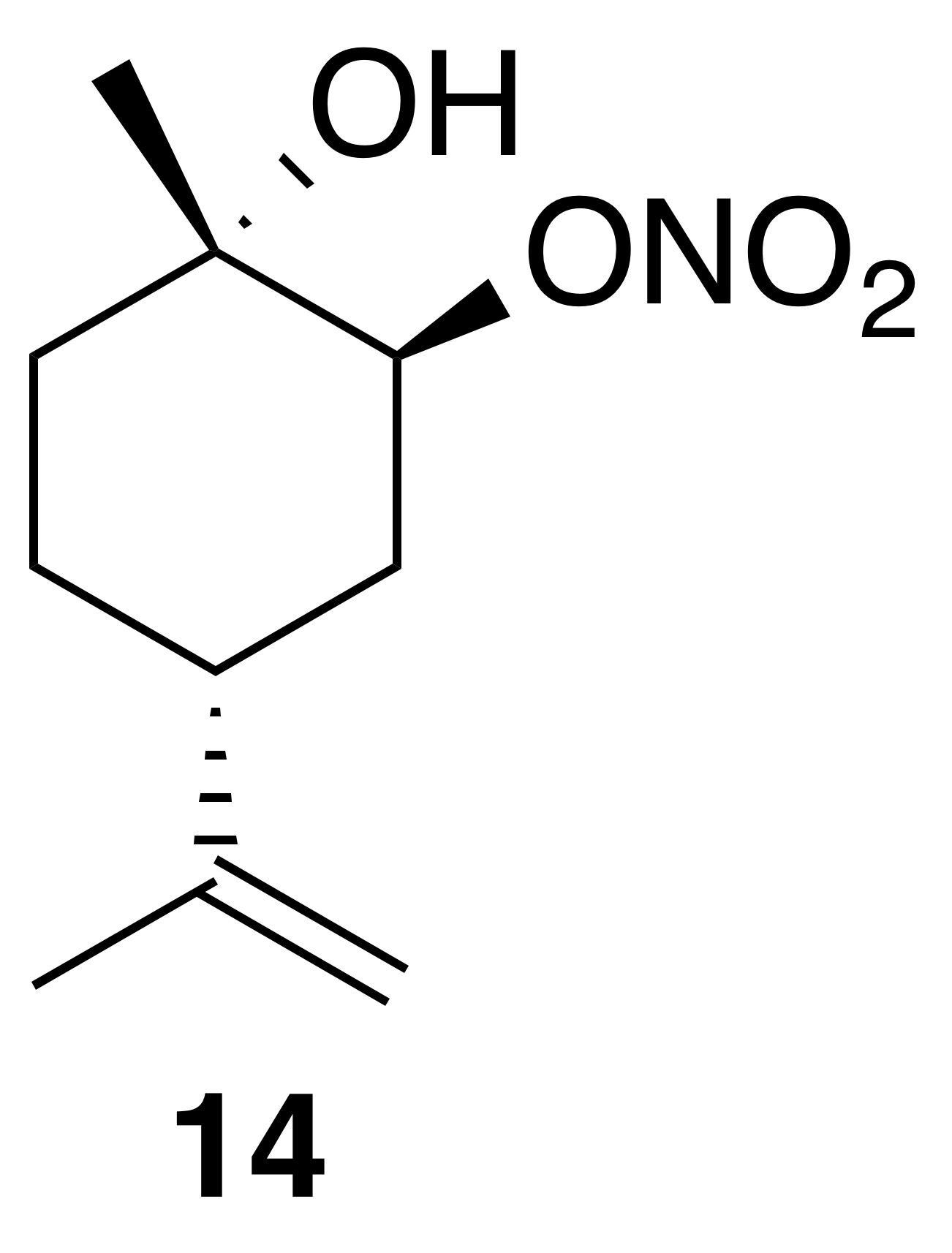
Nitration of trans-1,2-limonene oxide was carried out according to the general method with dioxane or dichloromethane. Purification by column chromatography (12 g SiO2, 0 %–50 % EtOAc ∕ Hex over 30 CVs) yielded nitrate ester (14) as a clear, colorless oil (63 % in dioxane; 54 % in DCM): Rf 0.39 (15 % EtOAc : Hex; anisaldehyde); IR (ATR) cm−1 3414 (bd w, alcoholic OH), 2940 (w), 1627 (s, NO2), 1279 (m, NO2), 876 (m, NO2), 851 (m, NO2); HRMS ESI + m∕z calculated for [C10H17O4NNa]+ (238.1048, observed; 238.1055, expected); 1H NMR (400 MHz, chloroform-d) δ 5.01 (s, 1 H), 4.76 (d, J=6.6 Hz, 2 H), 2.19 (tt, J=10.9, 3.1 Hz, 1 H), 2.03 (ddd, J=14.7, 12.1, 2.6 Hz, 1 H), 1.92 (dq, J=14.4, 3.5, 2.7 Hz, 1 H), 1.74 (s, 3 H), 1.69–1.56 (m, 4 H), 1.32 (s, 3 H); 13C NMR (101 MHz, chloroform-d) δ 148.27, 109.57, 84.43, 69.55, 37.96, 34.78, 29.91, 26.96, 20.89 ppm.
2.3.6 Nitration of perillic alcohol epoxide (15)
Nitration of 4-(1-methylethenyl)-7-oxabicyclo[4.1.0]heptane-1-methanol (15) was carried out according to the general method with dichloromethane. Purification by column chromatography (12 g SiO2, 0 %–40 % EtOAc ∕ Hex over 30 CVs) yielded two regioisomers and nitrate esters (16) and (17) in 23 % and 32 % yield, respectively.
Nitrate ester (16)
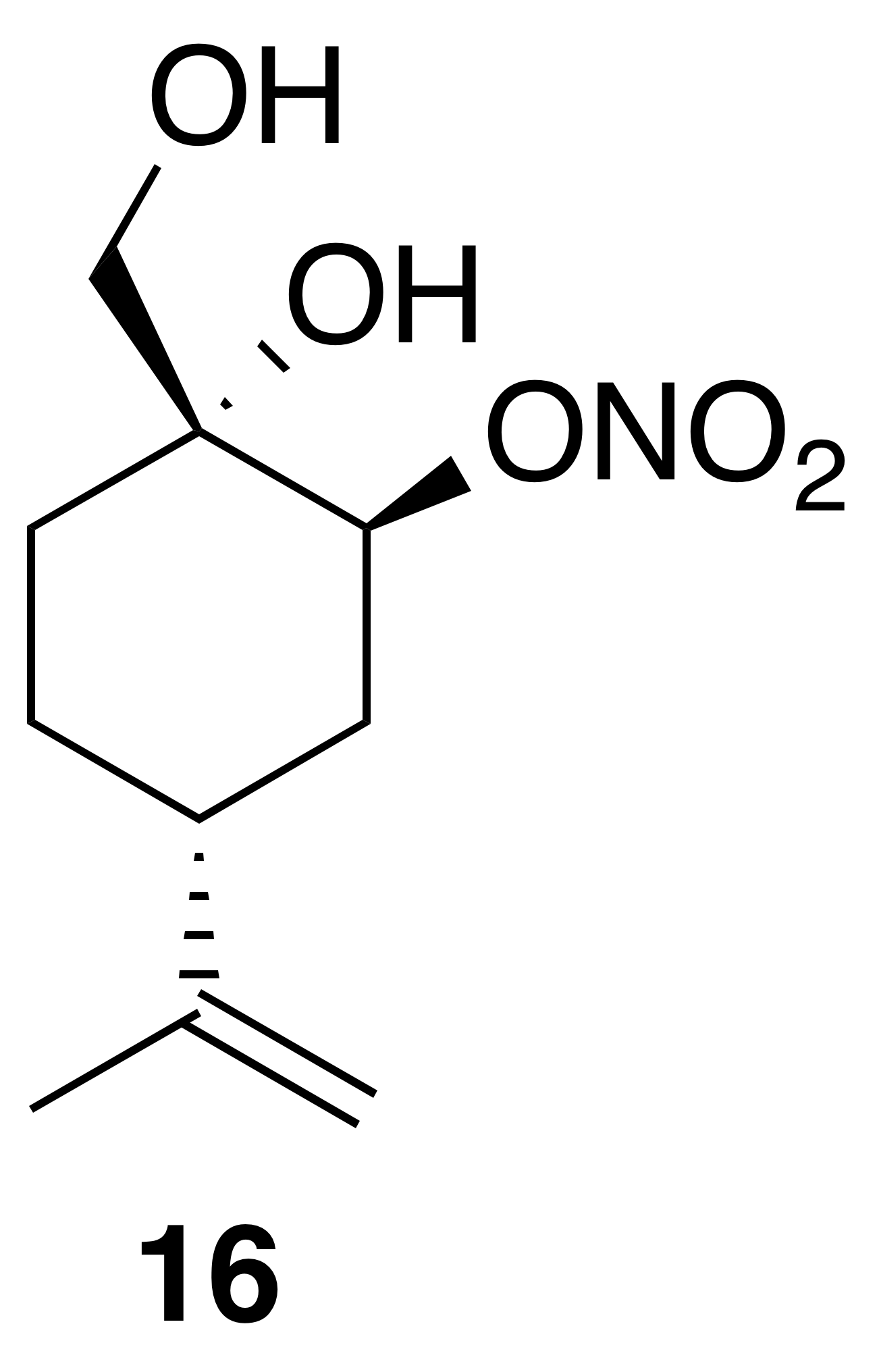
Nitrate ester (16) was isolated as a white, crystalline solid (106 mg, 23 % yield): mp = 72–75 ∘C; Rf 0.31 (30 % EtOAc : Hex; anisaldehyde); IR (ATR) cm−1 3285 (bd w, alcoholic OH), 2941 (w), 1627 (vs, NO2), 1280 (s, NO2), 880 (s, NO2), 850 (s, NO2); HRMS ESI + m∕z calculated for [C10H17O4NNa]+ (238.1054, observed; 238.1055, expected); 1H NMR (400 MHz, chloroform-d) δ 5.23 (s, 1 H), 4.80–4.73 (m, 2 H), 3.74 (dd, J=10.9, 5.0 Hz, 1 H), 3.44 (dd, J=11.0, 5.0 Hz, 1 H), 2.59 (d, J=1.0 Hz, 1 H), 2.27–2.16 (m, 1 H), 2.03–1.97 (m, 2 H), 1.79 (t, J=5.2 Hz, 1 H), 1.71–1.61 (m, 3 H), 1.58 (d, J=0.8 Hz, 4 H); 13C NMR (101 MHz, chloroform-d) δ 148.24, 109.58, 79.89, 70.98, 66.45, 38.47, 29.70, 29.55, 25.10, 20.77 ppm.
Nitrate ester (17)
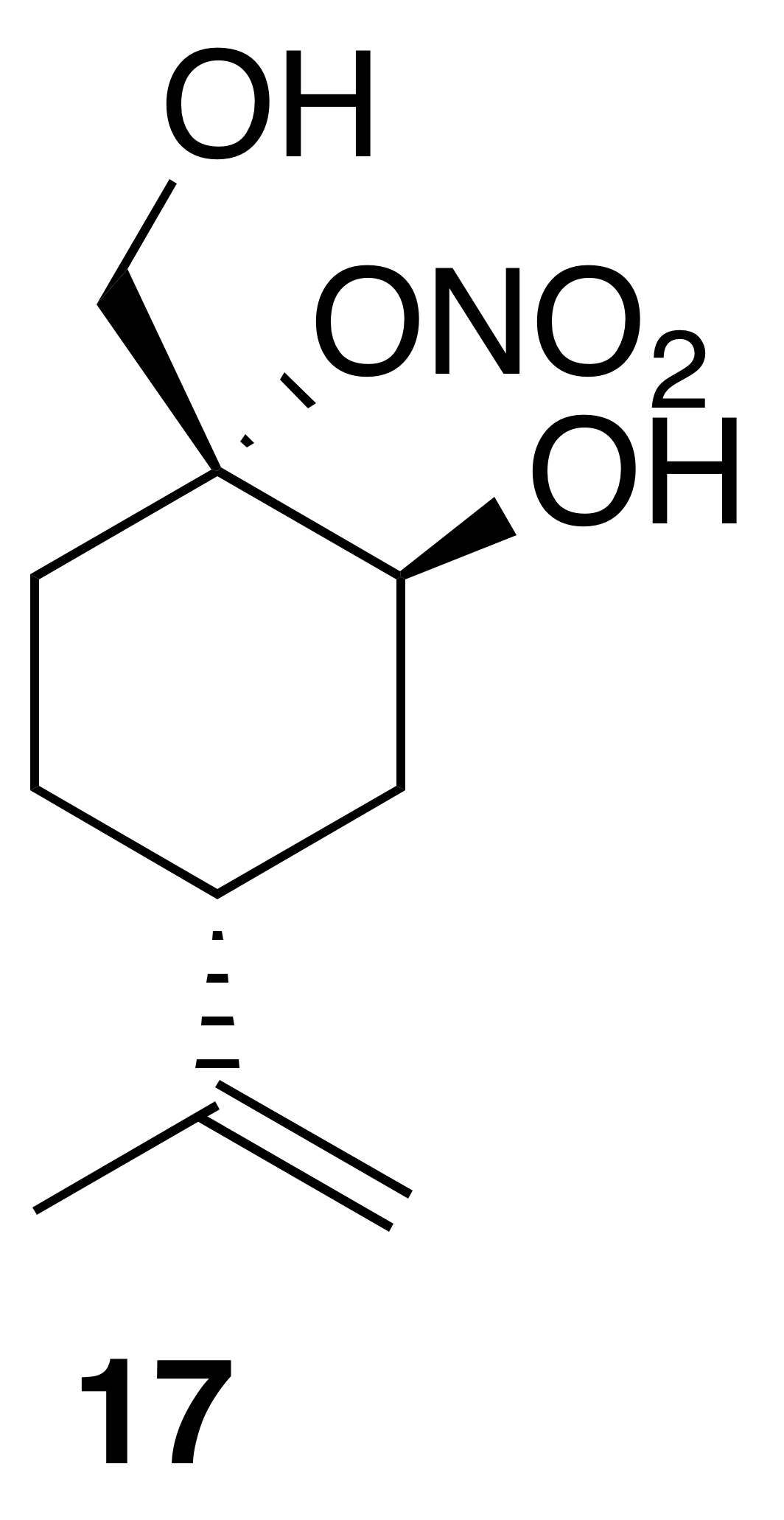
Nitrate ester (17) was isolated as a clear, colorless oil (149 mg, 32 % yield): Rf 0.20 (30 % EtOAc : Hex; anisaldehyde); IR (ATR) cm−1 3286 (bd w, alcoholic OH), 2941 (w), 1623 (vs, NO2), 1288 (s, NO2), 863 (vs, NO2); HRMS ESI − m∕z calculated for [C10H16O4N]− (214.10813, observed; 214.10848, expected); 1H NMR (400 MHz, chloroform-d) δ 4.76 (d, J=8.5 Hz, 2 H), 4.31 (s, 1 H), 4.27 (d, J=13.0 Hz, 1 H), 3.99 (d, J=13.0 Hz, 1 H), 2.41 (tt, J=12.1, 3.6 Hz, 1 H), 2.27–2.19 (m, 1 H), 1.98–1.78 (m, 3 H), 1.75 (s, 3 H), 1.72–1.65 (m, 1 H), 1.51–1.38 (m, 1 H); 13C NMR (101 MHz, chloroform-d) δ 148.58, 109.43, 92.51, 65.67, 62.91, 37.47, 33.53, 25.33, 25.26, 20.86 ppm.
2.3.7 Nitration of α-pinene oxide
Nitration of α-pinene oxide was carried out according to the general method. Crude oil from a 10 mmol reaction was purified by column chromatography (40 g SiO2, 0 %–40 % EtOAc ∕ Hex over 30 CVs) yielded the following isomerization products and nitrate esters.
α-Campholenic aldehyde (19)
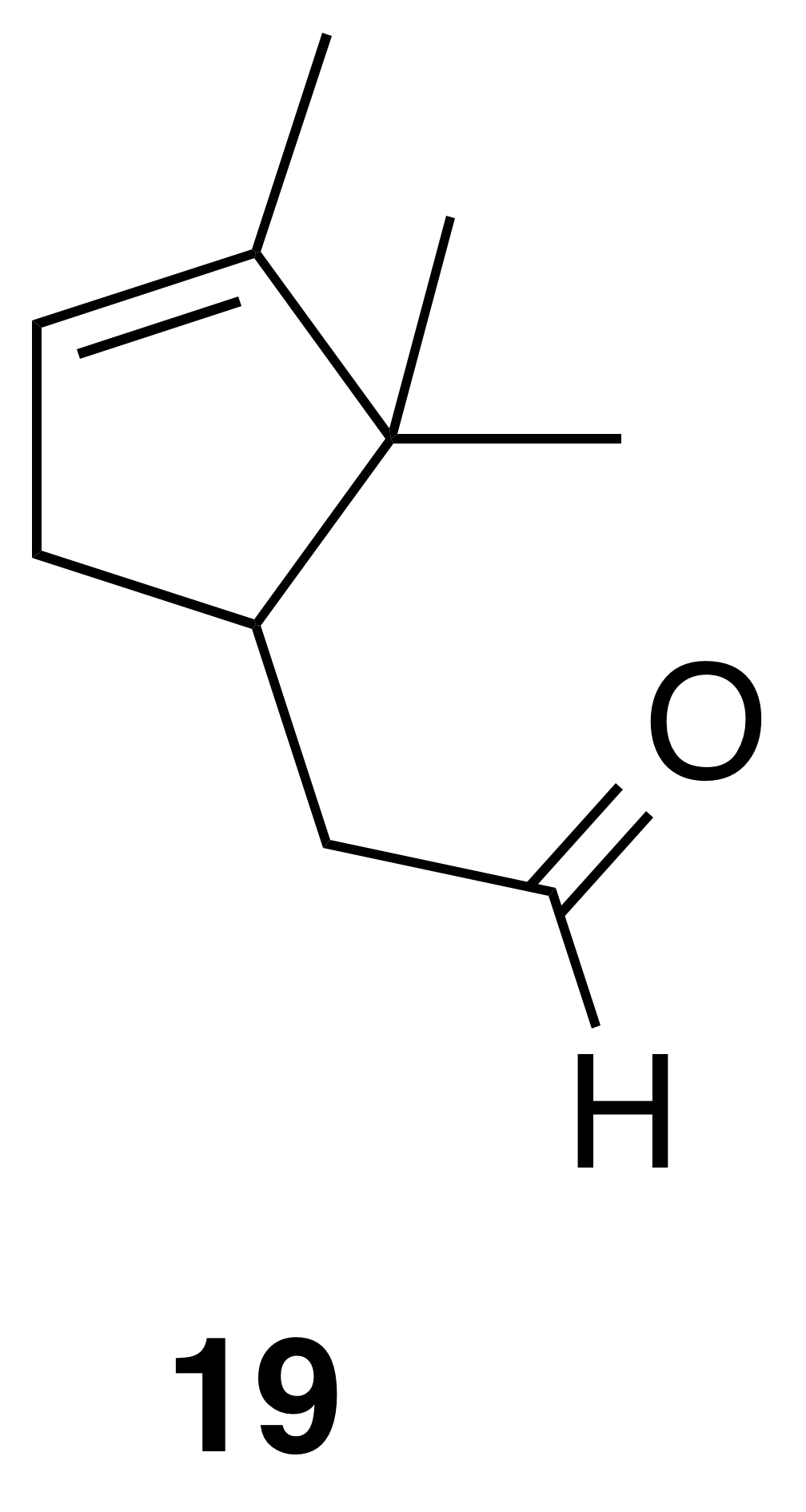
α-Campholenic aldehyde (19) was isolated as a clear, colorless oil: Rf 0.59 (10 % EtOAc : Hex; anisaldehyde); 1H NMR (400 MHz, benzene-d6) δ 9.51 (t, J=2.1 Hz, 1 H), 5.23 (s, 1 H), 2.41 (doublet of doublet of doublet of triplet (dddt), J=13.8, 6.0, 2.7, 1.6 Hz, 1 H), 2.25–2.16 (m, 1 H), 2.11 (ddd, J=15.9, 4.4, 1.8 Hz, 1 H), 2.00 (ddd, J=15.9, 10.3, 2.3 Hz, 1 H), 1.80 (doublet of doublet of pentet (ddp), J=15.8, 9.2, 2.5 Hz, 1 H), 1.58 (dt, J=3.0, 1.6 Hz, 3 H), 0.90 (s, 3 H), 0.68 (s, 3 H). Spectral data are consistent with literature reports (Thomas et al., 2016).
5-Isopropylidene-2-methyl-2-cyclohexen-1-ol (20)
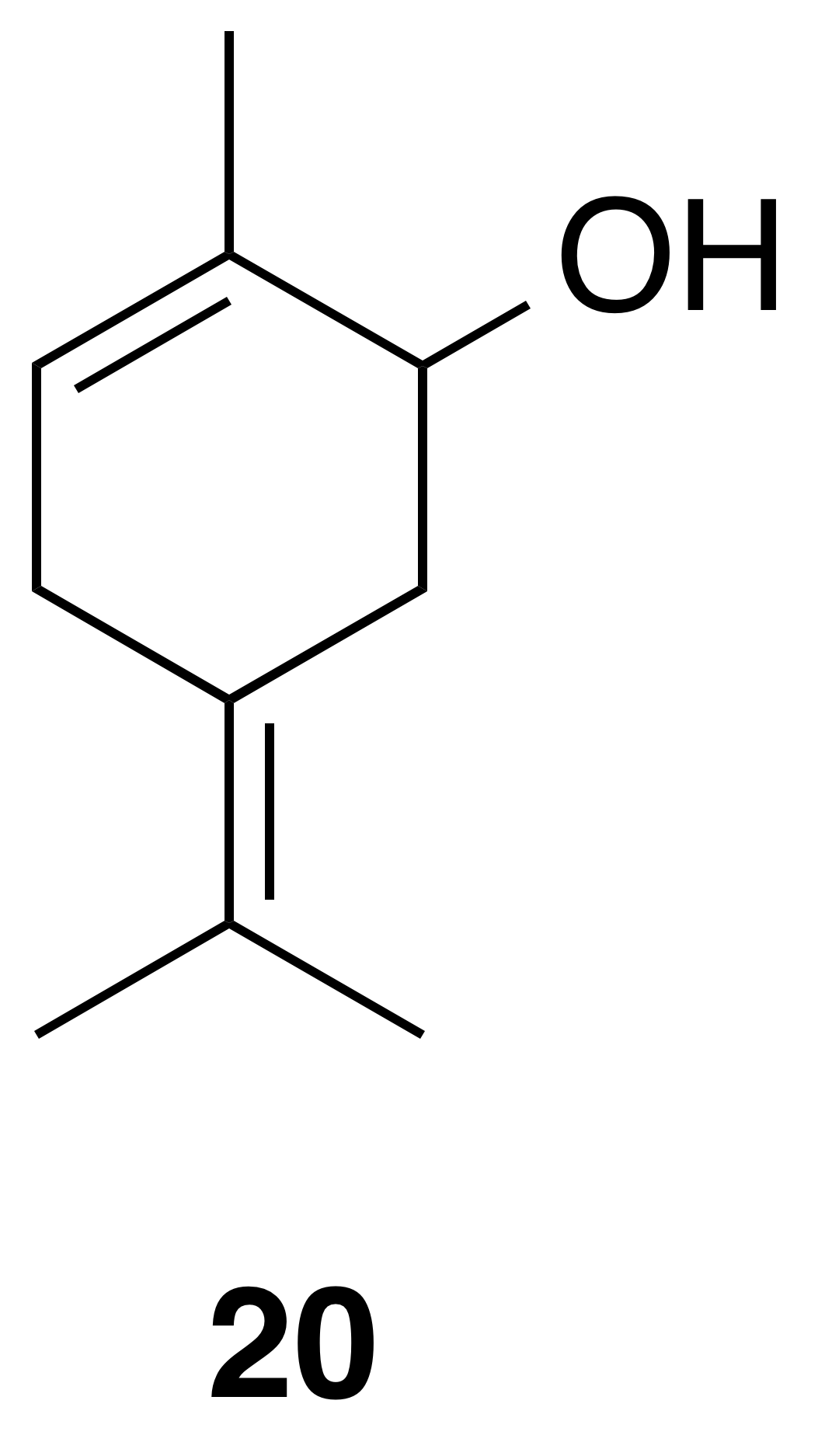
Compound (20) was isolated as a white, crystalline solid: Rf 0.47 (20 % EtOAc : Hex; anisaldehyde); 1H NMR (400 MHz, chloroform-d) δ 5.49 (ddd, J=4.4, 3.2, 1.5 Hz, 1 H), 3.99 (t, J=4.2 Hz, 1 H), 2.86 (d, J=19.6 Hz, 1 H), 2.69–2.56 (m, 2 H), 2.35 (d, J=15.5 Hz, 1 H), 1.78–1.75 (m, 3 H), 1.74 (s, 3 H), 1.68 (s, 3 H); 13C NMR (101 MHz, chloroform-d) δ 135.94, 125.57, 124.23, 123.29, 70.34, 35.92, 29.83, 20.31, 20.14, 19.91 ppm. Spectral data are consistent with literature reports (Motherwell et al., 2004).
trans-Carveol (21)
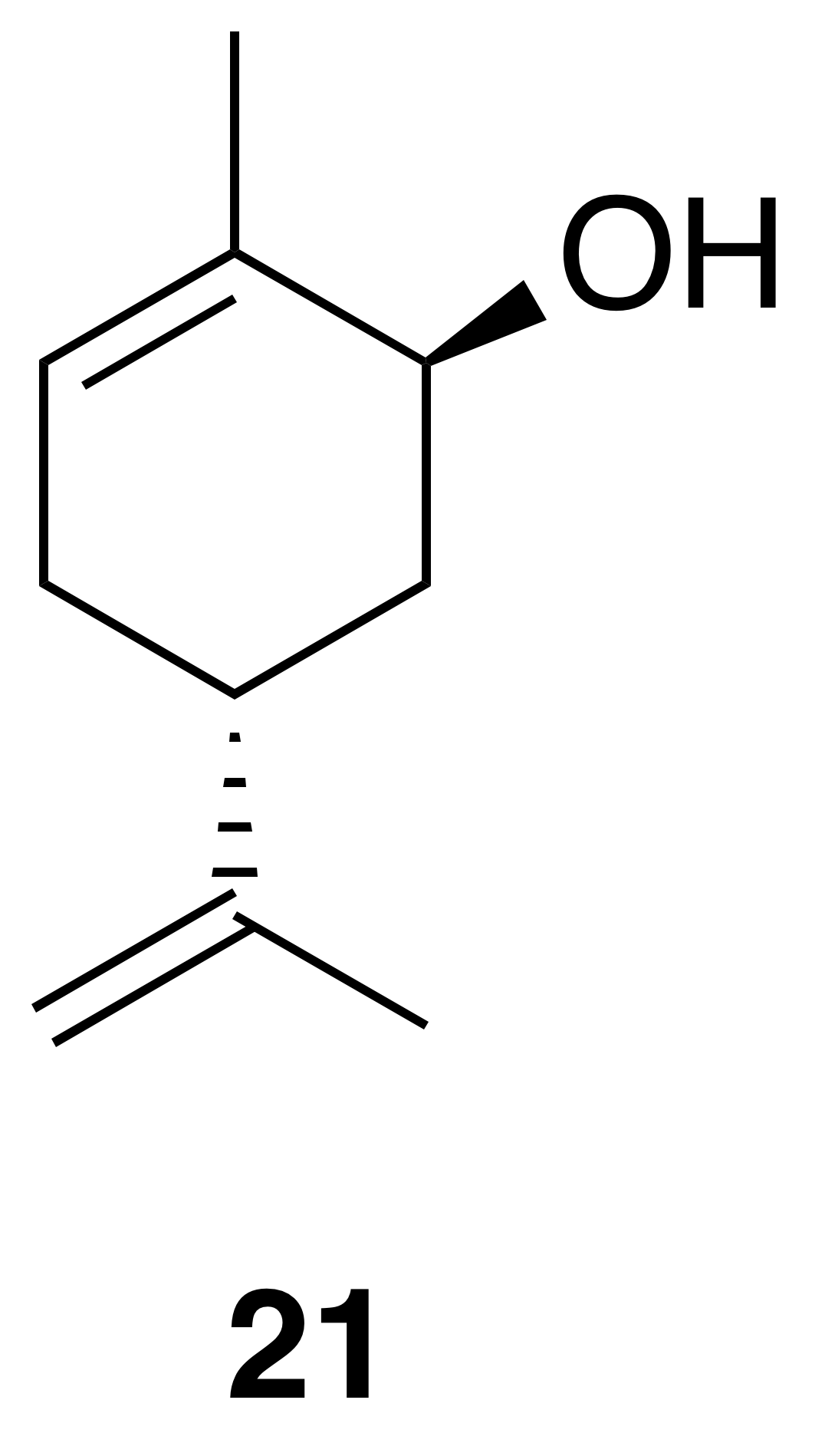
trans-Carveol was isolated as a clear, colorless oil: Rf 0.37 (20 % EtOAc : Hex; anisaldehyde); 1H NMR (400 MHz, chloroform-d) δ 5.62 (dd, J=5.2, 1.5 Hz, 1 H), 4.78–4.73 (m, 2 H), 4.06–4.03 (m, 1 H), 2.34 (ddd, J=13.7, 9.8, 2.9 Hz, 1 H), 2.21–2.12 (m, 1 H), 1.96 (dq, J=13.6, 2.3 Hz, 1 H), 1.93–1.85 (m, 1 H), 1.85–1.81 (m, 3 H), 1.77 (s, 3 H), 1.64 (dd, J=13.2, 4.0 Hz, 1 H), 1.61–1.57 (m, 1 H) ppm. Spectral data are consistent with literature reports) (Motherwell et al., 2004).
trans-Carveol nitrate ester (22)
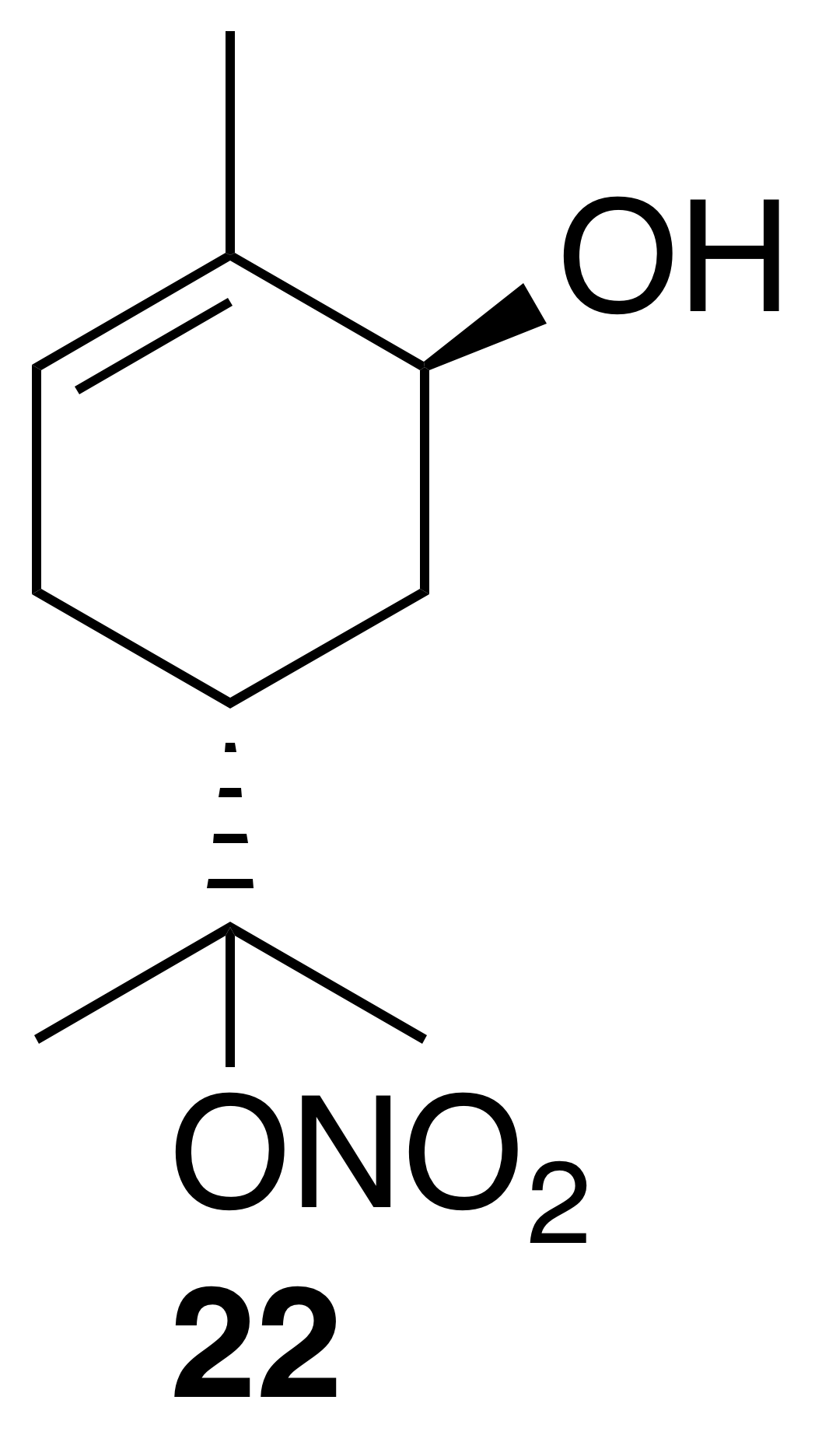
Nitrate ester (22) was isolated as a clear, colorless oil: IR (ATR) cm−1 3353 (bd w, alcoholic OH), 2917 (w), 1614 (vs, NO2), 1293 (s, NO2), 868 (m, NO2); 1H NMR (400 MHz, chloroform-d) δ 5.32 (ddd, J=9.9, 3.5, 2.1 Hz, 1 H), 4.12 (ddd, J=10.6, 4.6, 1.9 Hz, 1 H), 2.68–2.49 (m, 2 H), 1.88 (t, J=4.8 Hz, 1 H), 1.43 (dd, J=14.1, 3.7 Hz, 1 H), 1.24 (dd, J=13.8, 4.8 Hz, 1 H), 1.11 (s, 3 H), 0.97 (s, 3 H), 0.94 (s, 3 H) ppm. Spectral data are consistent with literature reports (Rindelaub et al., 2016b).
Nitrate ester (23)
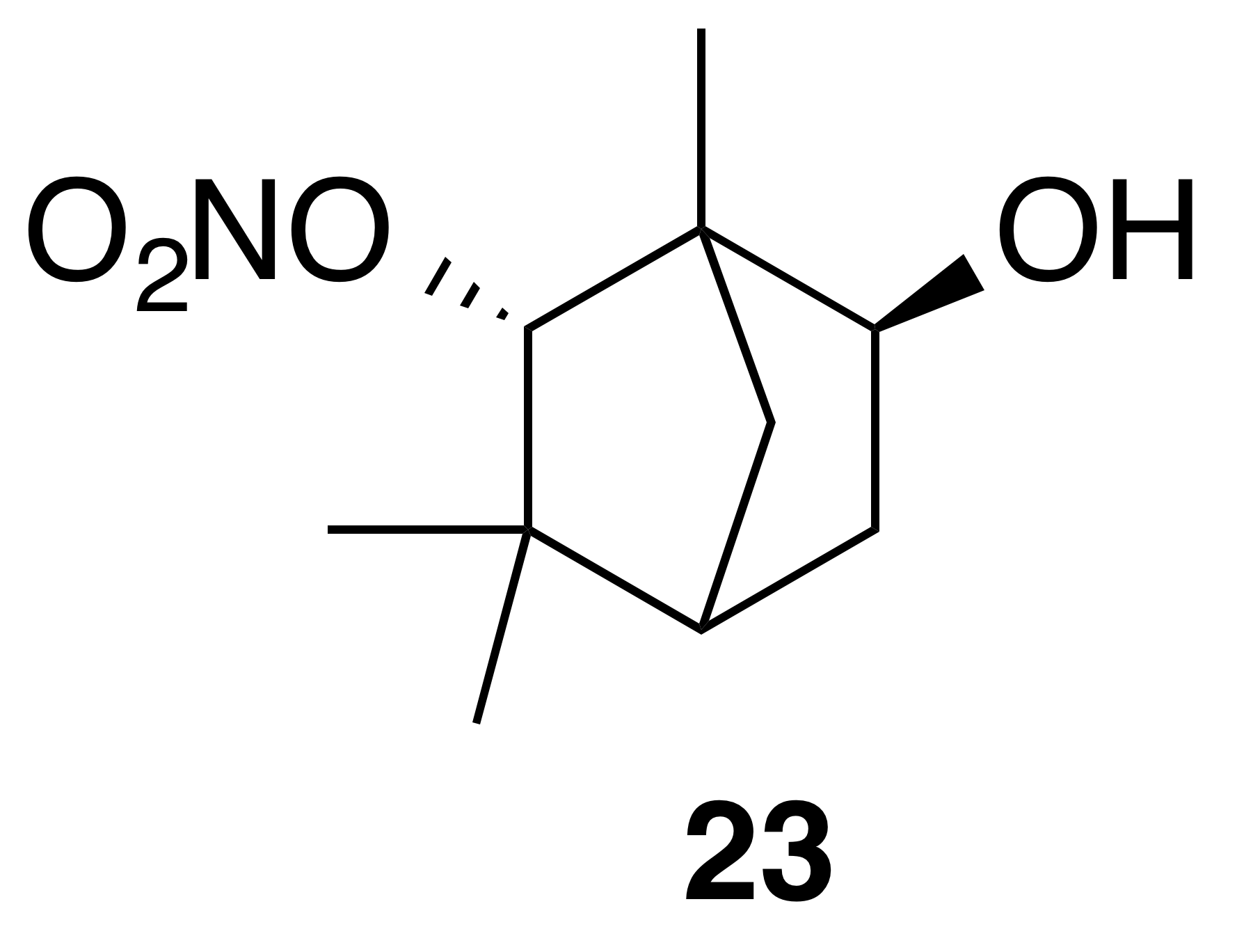
Nitrate ester (23) was isolated as a white, crystalline solid: mp = 132–134 ∘C; Rf 0.29 (25 % EtOAc : Hex; anisaldehyde); IR (ATR) cm−1 3294 (bd w, alcoholic OH), 2961 (w), 2360 (w), 1624 (m, NO2), 1282 (m, NO2), 862 (m, NO2); HRMS ESI − m∕z calculated for [C10H16O4N]− (214.10797, observed; 214.10848, expected); 1H NMR (400 MHz, chloroform-d) δ 4.64 (s, 1 H), 4.00 (ddd, J=7.1, 3.3, 0.9 Hz, 1 H), 2.37 (ddd, J=13.8, 7.1, 2.5 Hz, 1 H), 1.81 (d, J=4.3 Hz, 1 H), 1.60–1.51 (m, 3 H), 1.32 (dt, J=13.8, 4.0 Hz, 1 H), 1.23 (s, 3 H), 1.22 (s, 3 H), 0.89 (s, 3 H); 13C NMR (101 MHz, chloroform-d) δ 94.13, 68.52, 53.01, 47.34, 40.49, 37.76, 37.10, 29.24, 19.12, 14.75 ppm.
Nitrate ester (24)
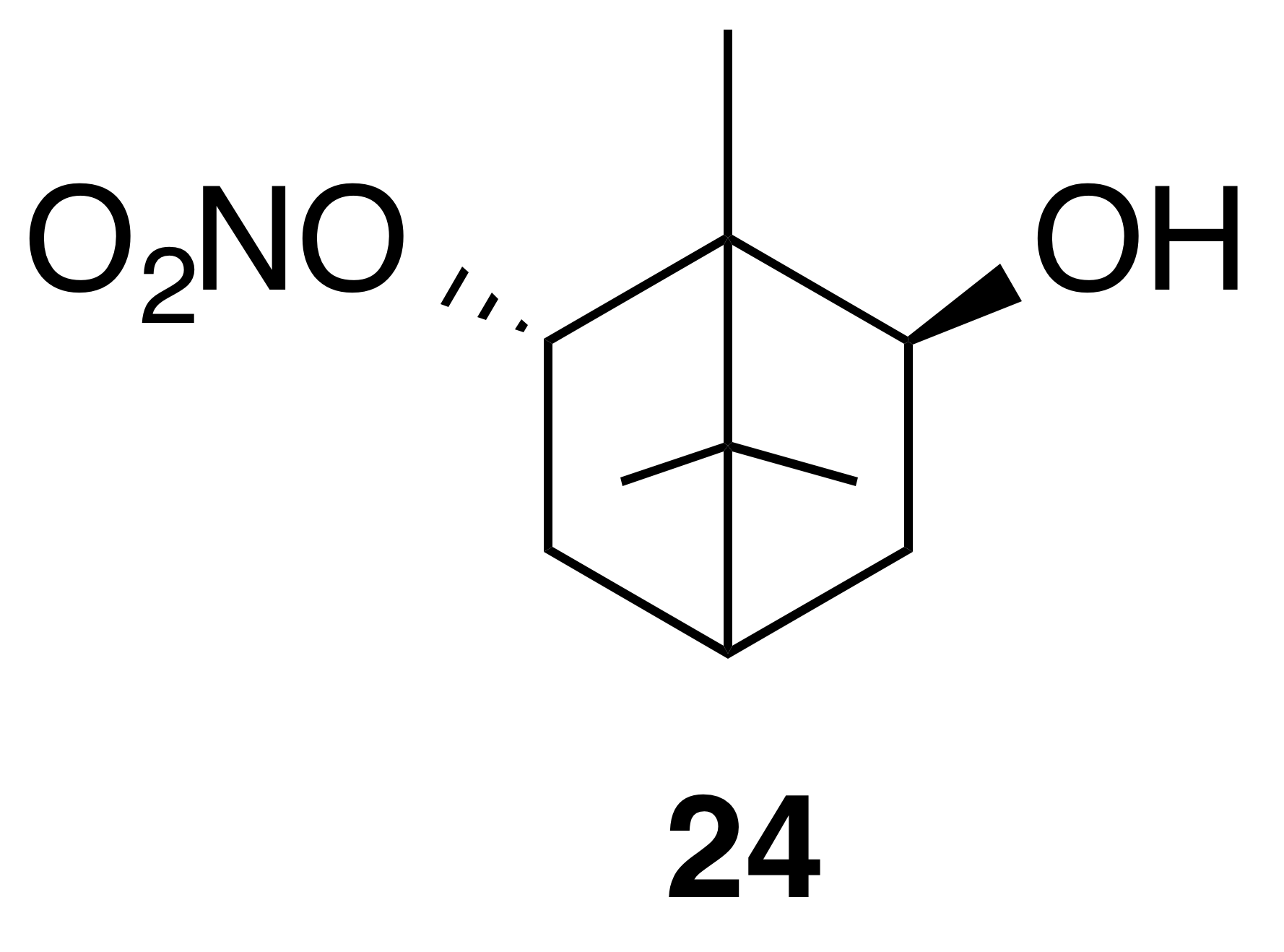
Nitrate ester (24) was isolated as a white, crystalline solid: mp = 62–64 ∘C; Rf 0.24 (15 % EtOAc : Hex; anisaldehyde); HRMS ESI + m∕z calculated for [C10H17O4NNa]+ (238.10459, observed; 238.10498, expected); IR (ATR) cm−1 3434 (w, alcoholic OH), 2959 (w), 1622 (s, NO2), 1284 (vs, NO2), 856 (m, NO2); 1H NMR (400 MHz, chloroform-d) δ 5.31 (ddd, J=9.9, 3.8, 2.1 Hz, 1 H), 4.12 (m, 1 H), 2.69–2.48 (m, 2 H), 1.87 (t, J=4.9 Hz, 1 H), 1.43 (dd, J=14.0, 3.7 Hz, 1 H), 1.24 (dd, J=13.7, 4.7 Hz, 1 H), 1.11 (s, 3 H), 0.97 (s, 3 H), 0.94 (s, 3 H); 13C NMR (101 MHz, chloroform-d) δ 91.04, 78.09, 52.58, 49.53, 42.73, 39.05, 36.71, 20.20, 19.70, 11.79 ppm.
2.3.8 Nitration of β-pinene oxide
Nitration of β-pinene oxide (11.1 mmol) was carried out according to the general procedure in toluene. Purification by column chromatography (80 g SiO2, 0 %–30 % EtOAc ∕ Hex over 20 CVs) yielded a 1 : 1 mix of myrtenol (26) and nitrate (29) (344 mg, 9 % combined yield), perillic alcohol (28, 600 mg, 36 % yield) and nitrate (27) (71 mg, 3 % yield). The combined myrtenol and nitrate (29) were separated by column chromatography (40 g SiO2; 5 %–20 % EtOAc ∕ Hex over 25 CVs).
Myrtenol (26)
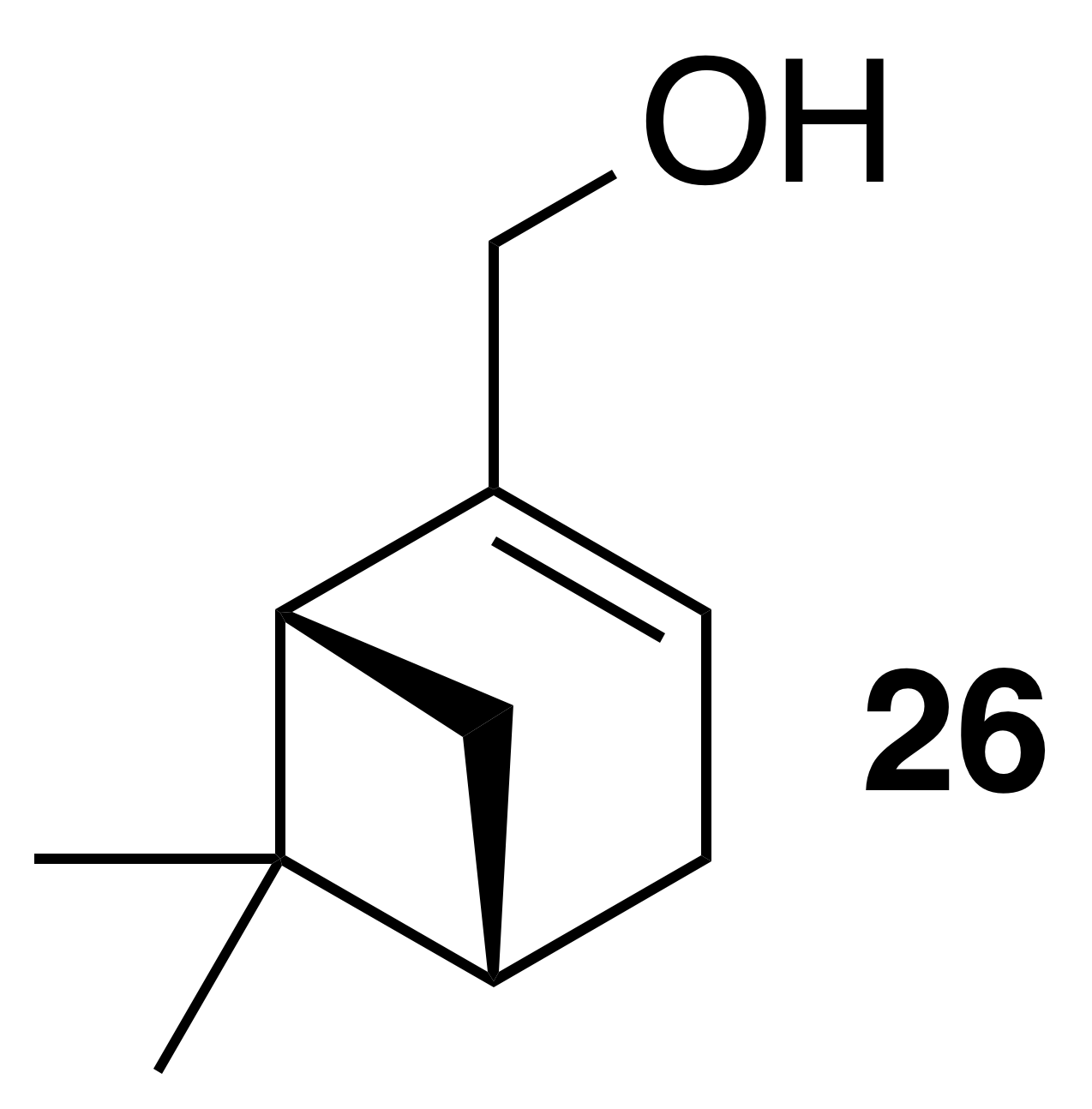
Myrtenol was isolated as a clear, colorless oil: Rf 0.25 (15 % EtOAc : Hex; anisaldehyde); 1H NMR (400 MHz, chloroform-d) δ 5.49 (dt, J=3.0, 1.5 Hz, 1 H), 4.00 (t, J=1.8 Hz, 2 H), 2.42 (dt, J=8.6, 5.6 Hz, 1 H), 2.37–2.20 (m, 2 H), 2.19–2.09 (m, 2 H), 1.61–1.45 (m, 1 H), 1.31 (s, 3 H), 1.19 (d, J=8.6 Hz, 1 H), 0.85 (s, 3 H) ppm. Spectral data are consistent with literature reports (Motherwell et al., 2004).
Nitrate ester (27)
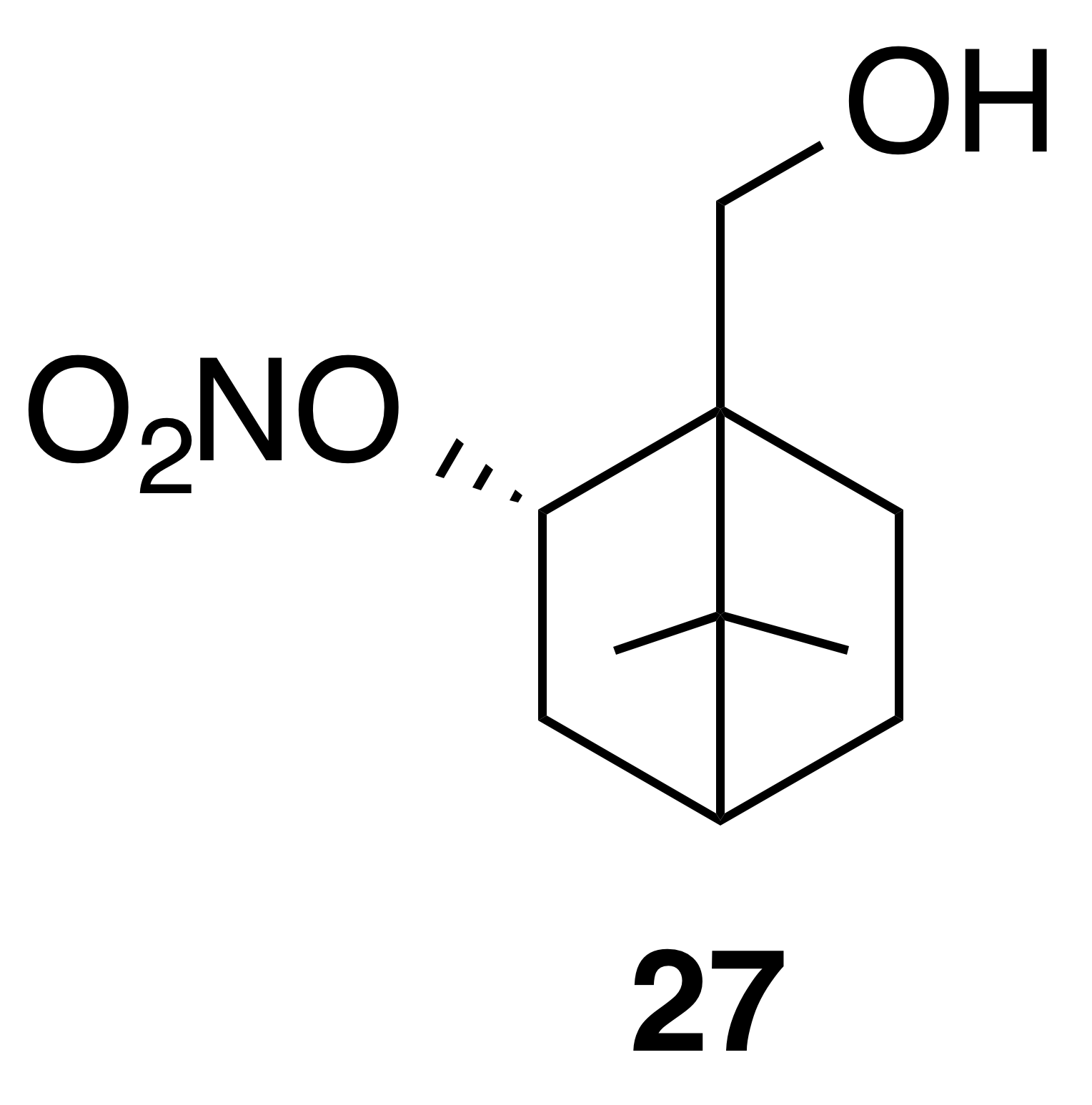
Nitrate ester (27) was isolated as a clear colorless oil: IR (ATR) cm−1 3362 (bd w, alcoholic OH), 2947 (w), 1624 (s, NO2), 1281 (vs, NO2), 864 (m, NO2); HRMS ESI − m∕z calculated for [C10H16O4N]− (214.10813, observed; 214.10848, expected); 1H NMR (400 MHz, chloroform-d) δ 5.57–5.49 (m, 1 H), 3.71 (q, J=11.5, 8.5 Hz, 2 H), 2.55 (ddt, J=14.1, 9.8, 3.9 Hz, 1 H), 1.95–1.83 (m, 2 H), 1.76 (t, J=4.5 Hz, 1 H), 1.52–1.22 (m, 5 H), 1.10 (s, 3 H), 1.01 (s, 3 H); 13C NMR (101 MHz, chloroform-d) δ 85.30, 62.08, 53.45, 48.62, 45.59, 35.78, 27.53, 22.88, 20.20, 20.02 ppm.
Nitrate ester (29)
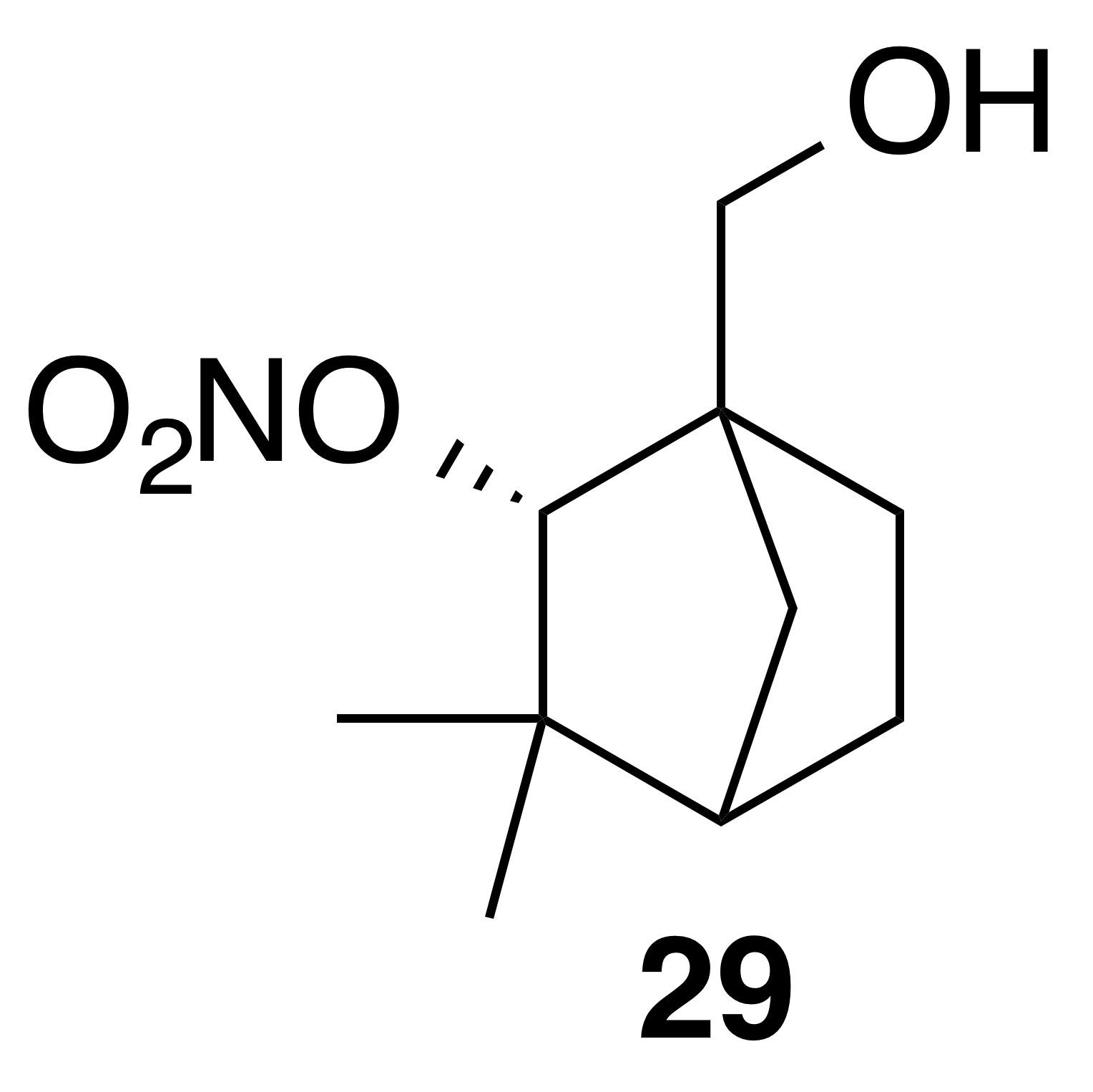
Nitrate ester (29) was isolated as a white, crystalline solid: mp = 59–61 ∘C; Rf 0.55 (15 % EtOAc : Hex; anisaldehyde); IR (ATR) cm−1 3374 (bd w, alcoholic OH), 2915 (w), 1625 (s, NO2), 1281 (vs, NO2), 863 (m, NO2); HRMS ESI + m∕z calculated for [C10H17O4NNa]+ (238.10474, observed; 238.10498, expected); 1H NMR (400 MHz, chloroform-d) δ 4.92 (d, J=1.6 Hz, 1 H), 3.75 (d, J=11.3 Hz, 1 H), 3.65 (d, J=11.3 Hz, 1 H), 1.90–1.80 (m, 3 H), 1.72–1.63 (m, 1 H), 1.56–1.50 (m, 1 H), 1.24–1.20 (m, 4 H), 0.96 (s, 3 H); 13C NMR (101 MHz, chloroform-d) δ 89.49, 63.82, 54.38, 48.25, 40.28, 37.03, 29.26, 24.74, 22.11, 19.46 ppm.
3.1 Synthesis
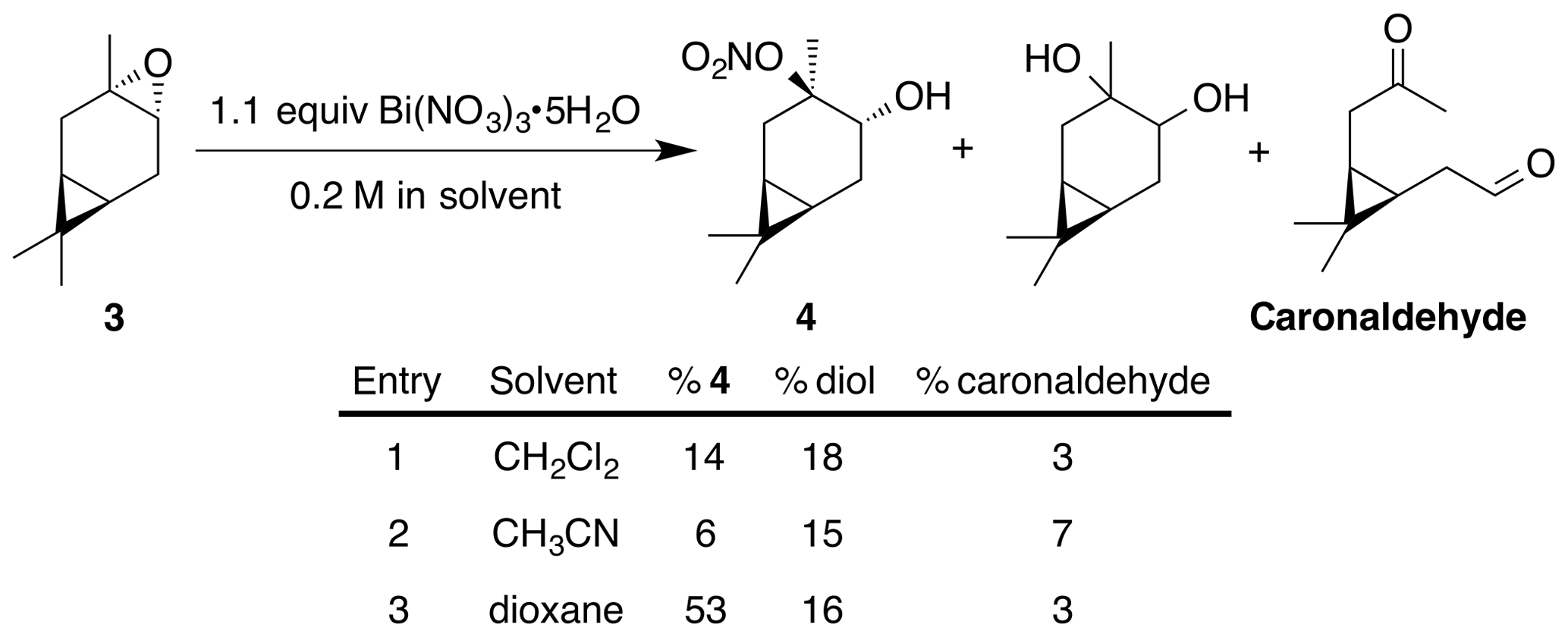
Previous reports have described the opening of epoxides and aziridines using bismuth nitrate in acetonitrile (Das et al., 2007). Others report optimal conditions in dichloromethane (Rindelaub et al., 2016b), or 1,4-dioxane with undesired side reactions in acetonitrile (Pinto et al., 2007). Thus, we began our investigations into the nitration of trans-carene oxide (3) using bismuth nitrate in various solvents (Fig. 2). In our hands, dioxane clearly outperformed dichloromethane, yielding 53 % of the desired product after 45 min (entry 3). A diol was isolated as a major (16 %) byproduct. Attempts to mitigate hydrolysis by the use of base or molecular sieves were ineffective. Acetonitrile produced a complex intractable mixture with a high amount of diol and caronaldehyde (entry 2). Other solvents, such as tetrahydrofuran (THF) and nitromethane, also produced complex mixtures with no trace of desired nitrate. Methanol, interestingly, only produced an undesired methyl ether. It is unclear if this product was generated through methanolysis of the nitrate ester or direct substitution of the epoxide. The regiochemistry of the nitration was easily elucidated with gHSQC NMR data. A doublet of doublets at 3.69 ppm correlated with a carbon shift at 71.1 ppm and was thus assigned to the alcohol methyne carbon. A tetrasubstituted carbon, with no correlations in the gHSQC at 95.6 ppm was consistent with the tertiary nitrate ester. Finally, the IR of compound (4) displayed the expected strong absorbances at 1616, 1291 and 868 cm−1.
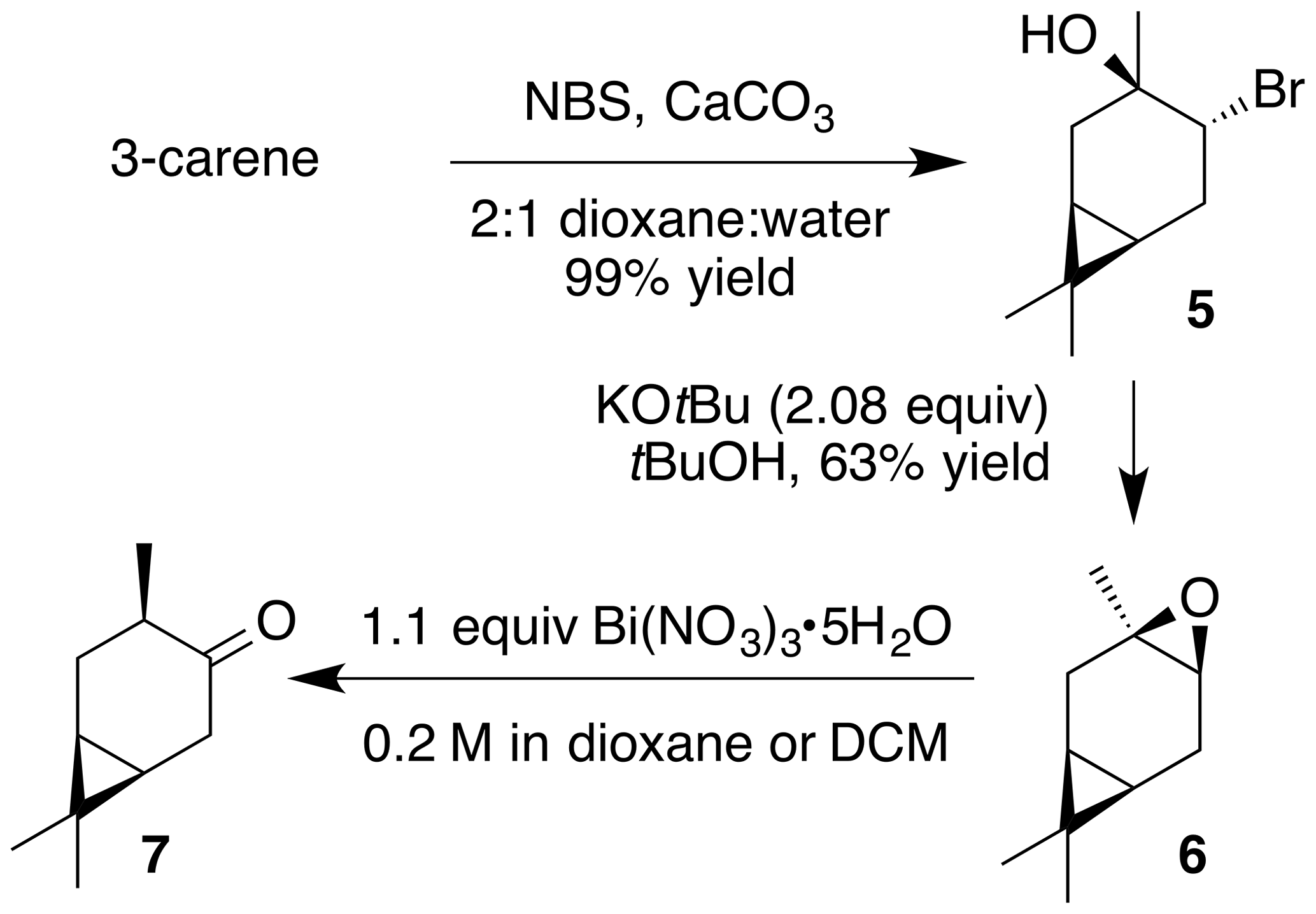
Next, we aimed to investigate the impact of relative stereochemistry on the nitration. We hypothesized that the secondary nitrate would predominate due to classical stereoelectronic effects. (+)-3-Carene was treated with N-bromosuccinimide and calcium carbonate to produce a bromohydrin which subsequently cyclized under basic conditions to produce cis-carene oxide (Cocker and Grayson, 1969). Interestingly, we never observed the desired nitrate ester. Instead a 1,2-hydrogen shift generated cis-4-caranone (7) in 53 % yield.
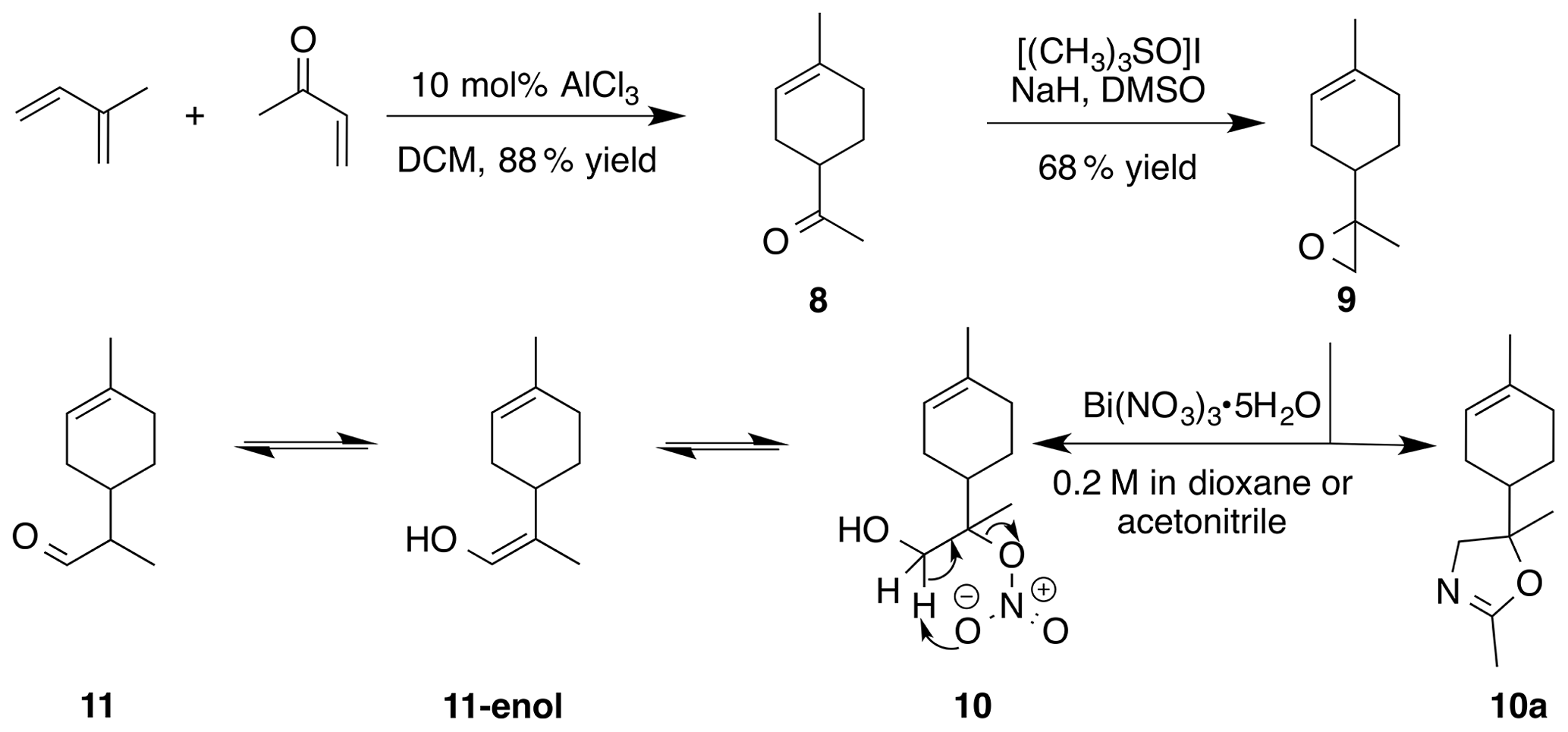
The preparation of 8,9-limonene oxide (9) began with a Diels–Alder reaction to form 1-(4-methyl-3-cyclohexene) ethenone (8) in 88 % yield. Corey–Chaykovsky addition of a sulfur ylide cleanly produced 8,9-limonene oxide (9). Nitration of (9) was attempted in a variety of solvents (DCM, benzene, dioxane, acetonitrile, nitromethane). In acetonitrile, oxazoline (10a) (1 : 1 d.r.) was cleanly produced presumably via nucleophilic addition of acetonitrile and subsequent cyclization to the oxazoline. Pinto et al. (2007) observed a similar bismuth nitrate mediated epoxide opening with acetonitrile. In dioxane, aldehyde (11) (1 : 1 d.r.) was observed along with a product consistent with a tertiary nitrate ester (10) (1 : 1 d.r.) in a 0.25 : 1 mixture. Formation of 11-enol presumably proceeds via a facile intramolecular elimination of nitric acid. The IR of the crude mixture showed the expected strong absorbances at 1613, 1288 and 865 cm−1. A set of signals in the 13C NMR at 97.40 and 97.17 ppm, with no gHSQC correlations, is consistent with a tertiary nitrate. Attempts to purify this mixture on silica gel gave small amounts of (11) and unidentifiable decomposition products. Furthermore, the fast hydrolysis rates of tertiary nitrate esters hinder the ability to isolate these products (Boyd et al., 2015; Darer et al., 2011; Liu et al., 2012). No reaction was observed when non-polar solvents such as DCM and benzene were employed. We hypothesize that the steric hindrance at the β-carbon severely limits reactivity.

Commercially available 1,2-limonene oxide was resolved into pure samples of cis- and trans-1,2-limonene oxide by treating with cyclic amine bases (Steiner et al., 2002). The nitration of the cis isomer was predicted to produce the tertiary nitrate ester via stereoelectronic effects. The dominant conformation of cis-1,2-limonene oxide is expected to be a pseudo-half chair (Fig. 5). Nucleophilic substitution at the less substituted position would proceed through a lower-energy chair-like transition state, whereas attack at the more substituted carbon leads to a very unstable twist-boat. The nitration of cis-1,2-limonene oxide (cis-12) proceeded smoothly in both dioxane (53 %) and dichloromethane (63 %). The trans isomer was expected to produce the secondary nitrate ester. The nitration of trans-1,2-limonene oxide (trans-12) also proceeded smoothly in both dioxane (63 %) and dichloromethane (61 %). Contrary to previous reports (Romonosky et al., 2015), we did not observe the desired nitrate esters in acetonitrile. gHSQC NMR data were used to verify nitrate ester (13) was tertiary and (14) was secondary. For (13), a broad singlet at 4.12 ppm correlated with a carbon shift at 69.2 ppm and was thus assigned to the methyne adjacent to the alcohol. A tetrasubstituted carbon at 91.4 ppm was consistent with the tetrasubstituted nitrate ester. Similarly for (14), a broad triplet at 5.00 ppm correlated with a carbon shift at 84.45 ppm and was thus assigned to the methyne adjacent to the nitrate ester. A tetrasubstituted carbon at 69.5 ppm was consistent with the tertiary alcohol.
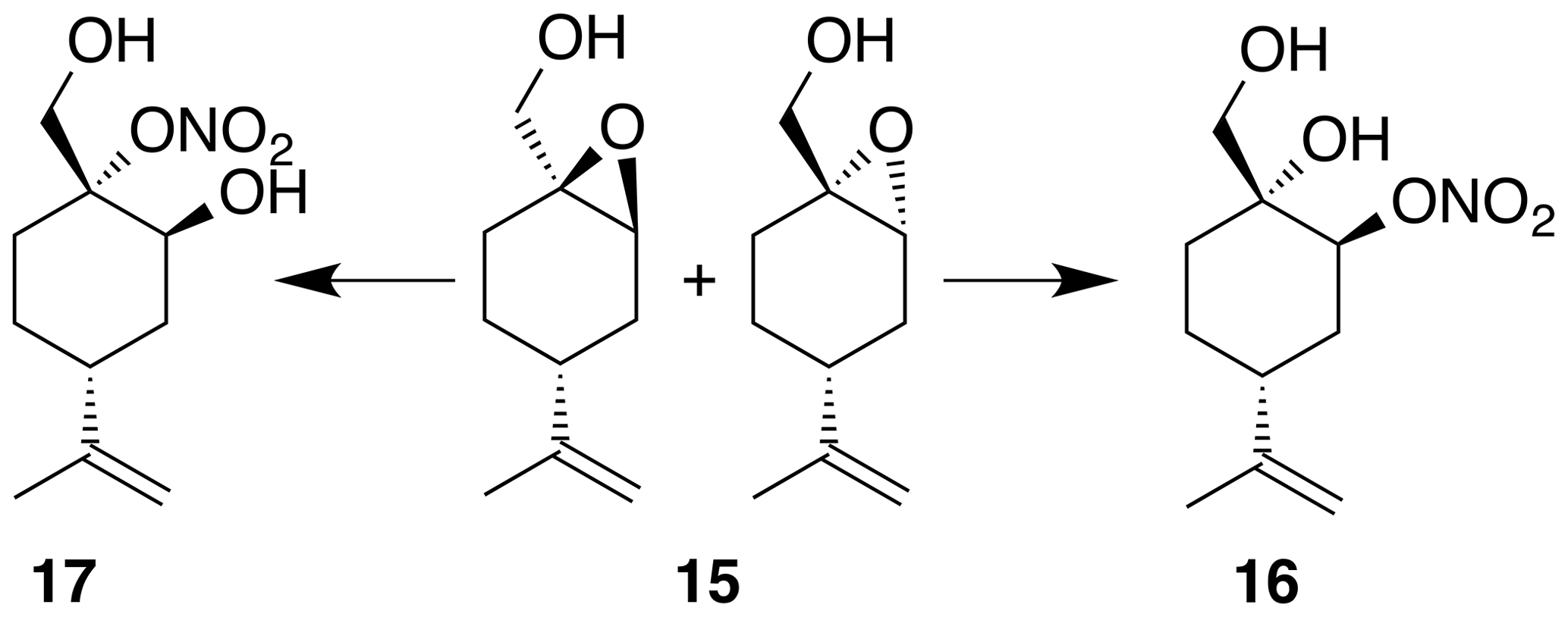
Under acidic conditions, β-pinene oxide undergoes facile rearrangement to form perillic alcohol. Accordingly, we prepared the epoxide (15) by treating perillic alcohol with one equivalent of m-CPBA. The inseparable mixture of diastereomers was treated with bismuth nitrate under the standard reaction conditions. Nitrate esters (16) and (17) were easily separated by column chromatography and assigned to the secondary and tertiary nitrate esters, respectively. These isomers were formed from the cis- and trans-epoxide diastereomers in an analogous fashion to the limonene isomers. For (16), a broad singlet at 5.21 ppm correlated with a carbon shift at 80.1 ppm and was thus assigned to the methyne adjacent to the nitrate ester. A tetrasubstituted carbon at 70.9 ppm was consistent with the tertiary alcohol. Similarly for (15), a broad singlet at 4.22 ppm correlated with a carbon shift at 65.6 ppm and was thus assigned to the methyne adjacent to the alcohol. A tetrasubstituted carbon at 92.8 ppm was consistent with the tertiary nitrate ester.

The facile rearrangements of α-pinene with both Brønsted and Lewis acids (Kaminska et al., 1992) can be thought to proceed via a nonclassical isobornyl cation (18) (Kong et al., 2010). We expected the reaction with bismuth nitrate to generate a complex mixture of products due to the many reactive sites. As shown in Fig. 7, three rearrangement products (campholenic aldehyde 19, diene 20 and trans-carveol 21) and three nitrate esters (22, 23, 24) were observed. The structure of nitrate ester (23) was identified by a clear singlet at 4.64 ppm (s, 1 H). Two-dimensional-NMR data (gCOSY, gHSQC and gHMBC) along with comparison to 6-exo-hydroxyfenchol, the analogous diol, confirmed the structural assignment (Miyazawa and Miyamoto, 2004). Correspondingly, nitrate ester (24) displayed a distinct doublet of doublets of doublets at 5.31 ppm that correlated to the methyne adjacent to the nitrate ester. Again, 2-D-NMR data (gCOSY, gHSQC and gHMBC) were used in conjunction with the literature spectra for platydiol and its trans diastereomer to verify the assignment (Kuo et al., 1989).
Under all conditions campholenic aldehyde was the major product (20 %–28 % yield). First generation nitrate ester (25) was not found under all conditions. All six components were isolated in small amounts when the reaction was run in dichloromethane (Fig. 7, entry 1). Cooling the reaction to −78 ∘C completely shut down all reactivity (entry 2). Interestingly, adding one equivalent of tetrabutylammonium nitrate (TBAN) as an external nitrate source in addition to bismuth nitrate at −78 ∘C generated mostly nitrate (22) (entry 3). More polar solvents, like dioxane (entry 4), generated slightly higher amounts of nitrates at room temperature. Aromatic solvents, such as benzene and toluene, provided the best yield of (23). Interestingly, adding TBAN to the nitration in toluene dramatically increased the amounts of diene (20) and trans-carveol (21). Using acetonitrile as a solvent generated a complex mixture that appeared to be mainly diols. Zirconium nitrate produced a similar distribution of products (Das et al., 2006), but other metal nitrate complexes (Y(NO3)3•6H2O, Co(NO3)2•6H2O) resulted in no observable reaction.
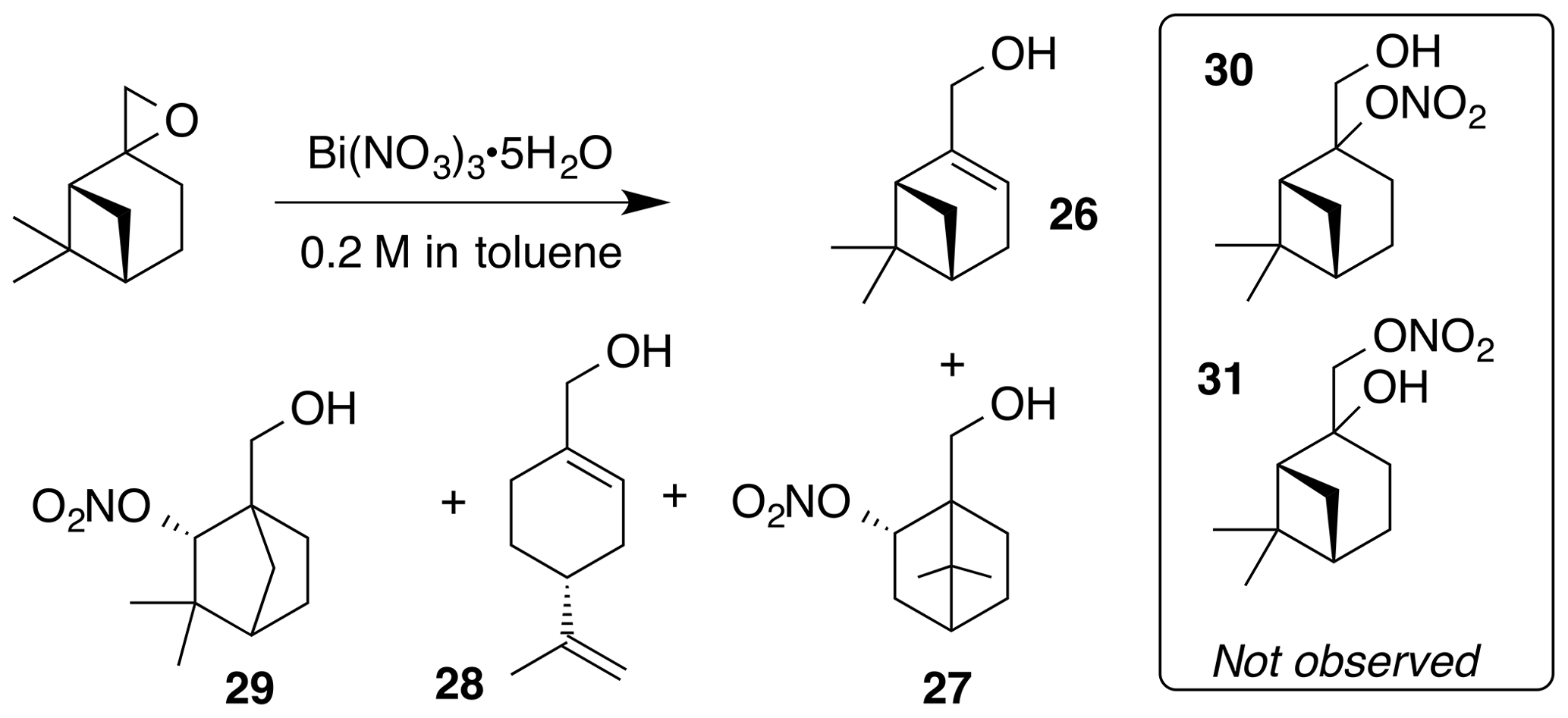
The nitration of β-pinene was similarly complicated by isomerization pathways through the corresponding non-classical carbocation (Fig. 8). Nitrate (27) initially co-eluted with myrtenol (26) but was separable upon a second purification by column chromatography. The methyne proton of (27) at 5.51 ppm is correlated with a carbon at 85.3 ppm in the gHSQC NMR. These data are consistent with the secondary nitrate ester. Similarly, the methyne proton in nitrate (29) is a doublet at 4.91 ppm (J4 = 1.6 Hz; W coupling). This signal correlates with a carbon at 89.5 ppm. Interestingly, nitrates (30) and (31) were not observed.
3.2 Spectral data
As shown in Table 1, all nitrate esters displayed the expected strong nitrate ester absorbances at ∼1600, 1300 and 900 cm−1. Methyne protons next to the secondary nitrate esters appeared between 4.64 and 5.51 ppm. Carbon chemical shifts were found over a broader range than anticipated from 80.1 to 95.6 ppm. Finally, we observed masses consistent with methanolysis products (185.1 m∕z M+; 223.1 m∕z M + K) when nitrate esters were analyzed (GC–MS) as solutions in methanol. Further experimentation is necessary to evaluate the implications of methanolysis.

3.3 Stability and storage
We were surprised to find that five of the nitrate esters (4, 16, 23, 24 and 29) are solids. We have stored these compounds at 0 ∘C for up to 9 months with no noticeable decline in purity. The remaining nitrate esters are stable at 0 ∘C for 2–4 weeks. Interestingly, some of these compounds appear to deviate from the expected stability patterns (vide supra). For example, tertiary nitrate ester (4) is particularly stable stored as a solid at 0 ∘C. In contrast, when secondary nitrate ester (14) was stored as a neat oil at 0 ∘C, it decomposed to a complex mixture within a few weeks. Freezing the samples as a solution in benzene is recommended for longer-term (∼6 months) storage.
We have clearly delineated successful methods required to synthesize and purify nine nitrate esters derived from mono-terpenes. Seven of these compounds are undescribed in the literature and the remaining two had gaps in their characterization. Using our methods it is possible to cleanly isolate 50–100 mg even of the least prevalent isomers from the nitration reactions of α-pinene and β-pinene. Interestingly, we did not observe the formation of α-pinene oxide and β-pinene oxide products that retained their bicyclic ring structures. This is consistent with the solution-phase synthetic literature (Kaminska et al., 1992) but in contrast to many atmospheric reports (for example, see Rindelaub et al., 2016b; Duporte et al., 2016). We believe that the availability of these compounds will enable further study of the structure–reactivity relationships. For example, comparing the specific hydrolysis rates for tertiary versus secondary nitrate esters in (13) and (14) as well as (17) and (16) could help deconvolute the fates of each terpene in the atmosphere. We also believe that these compounds will assist in confirming the identities of organic nitrates that have previously been limited to detection by MS-based methods (Rindelaub et al., 2016b). A forthcoming report will describe our investigation into the behavior of these authentic compounds in mass spectrometers. In particular, the detailed MS–MS data, impact of various ionization conditions (ESI, chemical ionization (CI), etc.) and sample preparation will be described. Finally, the availability of these compounds is important for further studies into the influence of terpene structure on the fate and roles of organic nitrates in SOA formation.
All spectral data for new compounds are available in the Supplement to this article.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-4241-2020-supplement.
RLL designed the experiments and prepared the manuscript with contributions from all co-authors. All co-authors carried out experiments and contributed to reviewing and editing the manuscript.
The authors declare that they have no conflict of interest.
This research was supported by generous start-up funding from Reed College. Elena Ali McKnight is grateful for funding from the Clark Fellowship. We thank Julie Fry for drawing our attention to this class of compounds. We are particularly grateful to Drew Gingerich, Nick Till, Stewart Green, Alyssa Harrison and Carlo Berti for preliminary experiments and to the 2014 and 2015 Chem 343 students for their assistance in this research.
This paper was edited by Jason Surratt and reviewed by five anonymous referees.
Almeida, Q. A. and Jones Jr., J.: Chemoselective Formation of 8,9-Epoxy-limonene, Synthetic Commun., 35, 1285–1290, https://doi.org/10.1081/scc-200057232, 2005.
Baker, J. W. and Easty, D. M.: Hydrolysis of organic nitrates, Nature, 166, p. 156, https://doi.org/10.1038/166156a0, 1950.
Bleier, D. B. and Elrod, M. J.: Kinetics and Thermodynamics of Atmospherically Relevant Aqueous Phase Reactions of α-Pinene Oxide, J. Phys. Chem., 117, 4223–4232, https://doi.org/10.1021/jp402093x, 2013.
Boyd, C. M., Sanchez, J., Xu, L., Eugene, A. J., Nah, T., Tuet, W. Y., Guzman, M. I., and Ng, N. L.: Secondary organic aerosol formation from the β-pinene + NO3 system: effect of humidity and peroxy radical fate, Atmos. Chem. Phys., 15, 7497–7522, https://doi.org/10.5194/acp-15-7497-2015, 2015.
Buss, A. D., Greeves, N., Mason, R., and Warren, S.: Applications of the stereochemically-controlled Horner-Wittig reaction: synthesis of feniculin, (E)-non-6-en-1-ol, a pheromone of the mediterranean fruit fly, (E)- and (Z)-dec-5-en-1-ol, tri-substituted alkenes, and (Z)-α-bisabolene, J. Chem. Soc. Perk. T. 1, 1987, 2569–2577, https://doi.org/10.1039/p19870002569, 1987.
Cabaj, J. E., Lukesh, J. M., Pariza, R. J., and Zizelman, P. M.: Large-Scale Preparation of (+)-p-Menth-2-ene-1,8-diol, a Key Intermediate in the Synthesis of Δ-9-Tetrahydrocannabinol, Org. Process Res. Dev., 1323, 358–361, https://doi.org/10.1021/op8002272, 2009.
Charbonneau, L., Foster, X., and Kaliaguine, S.: Ultrasonic and Catalyst-Free Epoxidation of Limonene and Other Terpenes Using Dimethyl Dioxirane in Semibatch Conditions, ACS Sustain. Chem. Eng., 6, 12224–12231, https://doi.org/10.1021/acssuschemeng.8b02578, 2018.
Cocker, W. and Grayson, D. H.: A convenient preparation of (−)-β-3,4-epoxycarane, Tetrahedron Lett., 10, 4451–4452, https://doi.org/10.1016/s0040-4039(01)88721-9, 1969.
Darer, A. I., Cole-Filipiak, N. C., O'Connor, A. E., and Elrod, M. J.: Formation and Stability of Atmospherically Relevant Isoprene-Derived Organosulfates and Organonitrates, Environ. Sci. Technol., 45, 1895–1902, https://doi.org/10.1021/es103797z, 2011.
Das, B., Krishnaiah, M., and Venkateswarlu, K.: Zirconyl nitrate mediated regioselective ring opening of epoxides and aziridines: an easy synthesis of β-nitrato-alcohols and -sulfonamides, Tetrahedron Lett., 47, 6027–6029, https://doi.org/10.1016/j.tetlet.2006.06.114, 2006.
Das, B., Krishnaiah, M., Venkateswarlu, K., and Reddy, V.: Efficient Regio- and Stereoselective Conversions of Oxiranes and Aziridines into β-(Nitrooxy)-Substituted Alcohols and Amines by Using Bismuth Nitrate, Helv. Chim. Acta, 90, 110–113, https://doi.org/10.1002/hlca.200790002, 2007.
Duporte, G., Flaud, P.-M., Geneste, E., Augagneur, S., Panui, E., Lamkaddam, H., Gratien, A., Doussin, J.-F., Budzinski, H., Villenave, E., and Perraudin, E.: Experimental Study of the Formation of Organosulfates from α-Pinene Oxidation. Part I: Product Identification, Formation Mechanisms and Effect of Relative Humidity, J. Phys. Chem. A, 120, 7909–7923, https://doi.org/10.1021/acs.jpca.6b08504, 2016.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hu, K. S., Darer, A. I., and Elrod, M. J.: Thermodynamics and kinetics of the hydrolysis of atmospherically relevant organonitrates and organosulfates, Atmos. Chem. Phys., 11, 8307–8320, https://doi.org/10.5194/acp-11-8307-2011, 2011.
Jacobs, M. I., Burke, W. J., and Elrod, M. J.: Kinetics of the reactions of isoprene-derived hydroxynitrates: gas phase epoxide formation and solution phase hydrolysis, Atmos. Chem. Phys., 14, 8933–8946, https://doi.org/10.5194/acp-14-8933-2014, 2014.
Kaminska, J., Schwegler, M., Hoefnagel, A., and van Bekkum, H.: The isomerization of α-pinene oxide with Brønsted and Lewis acids, Recl. Trav. Chim. Pay.-B., 111, 432–437, https://doi.org/10.1002/recl.19921111004, 1992.
Kolehmainen, E., Laihia, K., Heinänen, M., Rissanen, K., Fröhlich, R., Korvola, J., Mänttäri, P., and Kauppinen, R.: Oxygen-containing bicyclic monoterpenes. 1H, 13C and 17O NMR Spectroscopic and X-ray diffraction studies of seven oxidation products of (+)-3-carene, J. Chem. Soc. Perk. T., 2, 1993, 641–648, https://doi.org/10.1039/p29930000641, 1993.
Kong, J., Schleyer, P. v. R., and Rzepa, H. S.: Successful Computational Modeling of Isobornyl Chloride Ion-Pair Mechanisms, J. Org. Chem., 75, 5164–5169, https://doi.org/10.1021/jo100920e, 2010.
Kuo, Y.-H., Chen, W.-C., and Shih, K.-S.: ChemInform Abstract: Platydiol, a New Monoterpene from the Pericarp of Platycladus orientalis, Cheminform, 20, 511–514, https://doi.org/10.1002/chin.198946296, 1989.
Liu, S., Shilling, J. E., Song, C., Hiranuma, N., Zaveri, R. A., and Russell, L. M.: Hydrolysis of Organonitrate Functional Groups in Aerosol Particles, Aerosol. Sci. Tech., 46, 1359–1369, https://doi.org/10.1080/02786826.2012.716175, 2012.
Miyazawa, M. and Miyamoto, Y.: Biotransformation of (+)-(1R,2S)-fenchol by the larvae of common cutworm (Spodoptera litura), Tetrahedron, 60, 3091–3096, https://doi.org/10.1016/j.tet.2004.01.076, 2004.
Motherwell, W. B., Bingham, M. J., Pothier, J., and Six, Y.: A study of some molecularly imprinted polymers as protic catalysts for the isomerisation of α-pinene oxide to trans-carveol, Tetrahedron, 60, 3231–3241, https://doi.org/10.1016/j.tet.2004.02.016, 2004.
Nozière, B., Kalberer, M., Claeys, M., Allan, J., D'Anna, B., Decesari, S., Finessi, E., Glasius, M., Grgić, I., Hamilton, J. F., Hoffmann, T., Iinuma, Y., Jaoui, M., Kahnt, A., Kampf, C. J., Kourtchev, I., Maenhaut, W., Marsden, N., Saarikoski, S., Schnelle-Kreis, J., Surratt, J. D., Szidat, S., Szmigielski, R., and Wisthaler, A.: The Molecular Identification of Organic Compounds in the Atmosphere: State of the Art and Challenges, Chem. Rev., 115, 3919–3983, https://doi.org/10.1021/cr5003485, 2015.
Parker, K. A., Ledeboer, M. W., and Pettigrew, J. D.: Nitric Acid, in: Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd. Hoboken, NJ, https://doi.org/10.1002/047084289x.rn022.pub2, 1995.
Perring, A., Pusede, S., and Cohen, R.: An Observational Perspective on the Atmospheric Impacts of Alkyl and Multifunctional Nitrates on Ozone and Secondary Organic Aerosol, Chem. Rev., 113, 5848–5870, https://doi.org/10.1021/cr300520x, 2013.
Pinto, R., Salvador, J., and Roux, C.: Bismuth(III) salts mediated regioselective ring opening of epoxides: an easy route to halohydrins and β-hydroxy nitrates, Tetrahedron, 63, 9221–9228, https://doi.org/10.1016/j.tet.2007.06.054, 2007.
Pöschl, U., Carslaw, K., Koop, T., Sander, R., and Sturges, W.: Editorial Note “α-Pinene nitrates: synthesis, yields and atmospheric chemistry”' published in Atmos. Chem. Phys., 11, 6337–6347, 2011”, Atmos. Chem. Phys., 11, 12065–12065, https://doi.org/10.5194/acp-11-12065-2011, 2011.
Pye, H. O. T., Chan, A. W. H., Barkley, M. P., and Seinfeld, J. H.: Global modeling of organic aerosol: the importance of reactive nitrogen (NOx and NO3), Atmos. Chem. Phys., 10, 11261–11276, https://doi.org/10.5194/acp-10-11261-2010, 2010.
Reid, W. B. and Watson, D. A.: Synthesis of Trisubstituted Alkenyl Boronic Esters from Alkenes Using the Boryl-Heck Reaction, Org. Lett., 20, 6832–6835, https://doi.org/10.1021/acs.orglett.8b02949, 2018.
Rindelaub, J. D., McAvey, K. M., and Shepson, P. B.: Determination of α-pinene-derived organic nitrate yields: particle phase partitioning and hydrolysis, Atmos. Chem. Phys. Discuss., 14, 3301–3335, https://doi.org/10.5194/acpd-14-3301-2014, 2014.
Rindelaub, J. D., McAvey, K. M., and Shepson, P. B.: The photochemical production of organic nitrates from α-pinene and loss via acid-dependent particle phase hydrolysis, Atmos. Environ., 100, 193–201, https://doi.org/10.1016/j.atmosenv.2014.11.010, 2015.
Rindelaub, J. D., Wiley, J. S., Cooper, B. R., and Shepson, P. B.: Chemical characterization of α-pinene secondary organic aerosol constituents using gas chromatography, liquid chromatography, and paper spray-based mass spectrometry techniques, Rapid Commun. Mass Sp., 30, 1627–1638, https://doi.org/10.1002/rcm.7602, 2016a.
Rindelaub, J. D., Borca, C. H., Hostetler, M. A., Slade, J. H., Lipton, M. A., Slipchenko, L. V., and Shepson, P. B.: The acid-catalyzed hydrolysis of an α-pinene-derived organic nitrate: kinetics, products, reaction mechanisms, and atmospheric impact, Atmos. Chem. Phys., 16, 15425–15432, https://doi.org/10.5194/acp-16-15425-2016, 2016b.
Rollins, A. W., Fry, J. L., Hunter, J. F., Kroll, J. H., Worsnop, D. R., Singaram, S. W., and Cohen, R. C.: Elemental analysis of aerosol organic nitrates with electron ionization high-resolution mass spectrometry, Atmos. Meas. Tech., 3, 301–310, https://doi.org/10.5194/amt-3-301-2010, 2010.
Romonosky, D. E., Nguyen, L. Q., Shemesh, D., Nguyen, T. B., Epstein, S. A., Martin, D., Vanderwal, C. D., Gerber, B. R., and Nizkorodov, S. A.: Absorption spectra and aqueous photochemistry of β-hydroxyalkyl nitrates of atmospheric interest, Mol. Phys., 113, 2179–2190, https://doi.org/10.1080/00268976.2015.1017020, 2015.
Steiner, D., Ivison, L., Goralski, C. T., Appell, R. B., Gojkovic, J. R., and Singaram, B.: A facile and efficient method for the kinetic separation of commercially available cis- and trans-limonene epoxide, Tetrahedron Asymmetry, 13, 2359–2363, https://doi.org/10.1016/s0957-4166(02)00646-8, 2002.
Thomas, W. C., Dresser, W. D., Cortes, D. A., and Elrod, M. J.: Gas Phase Oxidation of Campholenic Aldehyde and Solution Phase Reactivity of its Epoxide Derivative, J. Phys. Chem., 121, 168–180, https://doi.org/10.1021/acs.jpca.6b08642, 2016.
Uehara, A., Tommis, B., Belhassen, E., Satrani, B., Ghanmi, M., and Baldovini, N.: Odor-active constituents of Cedrus atlantica wood essential oil, Phytochemistry, 144, 208–215, https://doi.org/10.1016/j.phytochem.2017.09.017, 2017.
Univ. of California, Berkeley: Lesson Learned – Nitric Acid Waste Over-pressurization Event, available at: https://ehs.berkeley.edu/lessons-learned/lesson-learned-nitric-acid-waste-over-pressurization-event, last access: 1 July 2019.