the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 20 Mar 2020
| 20 Mar 2020
Importance of isomerization reactions for OH radical regeneration from the photo-oxidation of isoprene investigated in the atmospheric simulation chamber SAPHIR
Luc Vereecken
Birger Bohn
Hans-Peter Dorn
Georgios I. Gkatzelis
Andreas Hofzumahaus
Frank Holland
David Reimer
Franz Rohrer
Simon Rosanka
Domenico Taraborrelli
Ralf Tillmann
Robert Wegener
Zhujun Yu
Astrid Kiendler-Scharr
Andreas Wahner
Hendrik Fuchs
Theoretical, laboratory, and chamber studies have shown fast regeneration of the hydroxyl radical (OH) in the photochemistry of isoprene, largely due to unimolecular reactions which were previously thought not to be important under atmospheric conditions. Based on early field measurements, nearly complete regeneration was hypothesized for a wide range of tropospheric conditions, including areas such as the rainforest where slow regeneration of OH radicals is expected due to low concentrations of nitric oxide (NO). In this work the OH regeneration in isoprene oxidation is directly quantified for the first time through experiments covering a wide range of atmospherically relevant NO levels (between 0.15 and 2 ppbv – parts per billion by volume) in the atmospheric simulation chamber SAPHIR. These conditions cover remote areas partially influenced by anthropogenic NO emissions, giving a regeneration efficiency of OH close to 1, and areas like the Amazonian rainforest with very low NO, resulting in a surprisingly high regeneration efficiency of 0.5, i.e. a factor of 2 to 3 higher than explainable in the absence of unimolecular reactions. The measured radical concentrations were compared to model calculations, and the best agreement was observed when at least 50 % of the total loss of isoprene peroxy radicals conformers (weighted by their abundance) occurs via isomerization reactions for NO lower than 0.2 ppbv. For these levels of NO, up to 50 % of the OH radicals are regenerated from the products of the 1,6 α-hydroxy-hydrogen shift (1,6-H shift) of Z-δ-RO2 radicals through the photolysis of an unsaturated hydroperoxy aldehyde (HPALD) and/or through the fast aldehydic hydrogen shift (rate constant ∼10 s−1 at 300 K) in di-hydroperoxy carbonyl peroxy radicals (di-HPCARP-RO2), depending on their relative yield. The agreement between all measured and modelled trace gases (hydroxyl, hydroperoxy, and organic peroxy radicals, carbon monoxide, and the sum of methyl vinyl ketone, methacrolein, and hydroxyl hydroperoxides) is nearly independent of the adopted yield of HPALD and di-HPCARP-RO2 as both degrade relatively fast (<1 h), forming the OH radical and CO among other products. Taking into consideration this and earlier isoprene studies, considerable uncertainties remain on the distribution of oxygenated products, which affect radical levels and organic aerosol downwind of unpolluted isoprene-dominated regions.
- Article
(5159 KB) -
Supplement
(2551 KB) - BibTeX
- EndNote





The hydroxyl radical (OH) is the main daytime oxidant controlling the removal and transformation of gaseous pollutants in the atmosphere (Levy, 1974). Its high efficiency in the oxidation of trace gases is based on the effective regeneration of OH by radical chain reactions, in which nitric oxide (NO) is oxidized to nitrogen dioxide (NO2), linking the OH chemistry to the formation of tropospheric pollutant ozone (O3). Because high levels of OH radicals were observed in field experiments in mainly forested environments with large concentrations of isoprene (Tan et al., 2001; Ren et al., 2008; Hofzumahaus et al., 2009; Kubistin et al., 2010; Whalley et al., 2011), a large number of investigations over the last decade focused on OH-initiated isoprene chemistry, including laboratory and chamber studies (Crounse et al., 2011; Berndt, 2012; Wolfe et al., 2012; Fuchs et al., 2013; Teng et al., 2017; Berndt et al., 2019), theoretical calculations (Peeters et al., 2009, 2014; Da Silva et al., 2010; Peeters and Müller, 2010; 2014; Wang et al., 2018; Møller et al., 2019), and global model impact (Lelieveld et al., 2008; Taraborrelli et al., 2012; Bates and Jacob, 2019; Møller et al., 2019; Müller et al., 2019). The observed OH levels could only be explained if an OH radical regeneration mechanism exists independently of NO and thus without the formation of O3. It is now widely accepted that unimolecular isomerization reactions of peroxy radicals (RO2) formed during the oxidation of organic compounds can contribute to the regeneration of radicals, in particular if they become competitive against RO2 radical losses via NO (Praske et al., 2018).
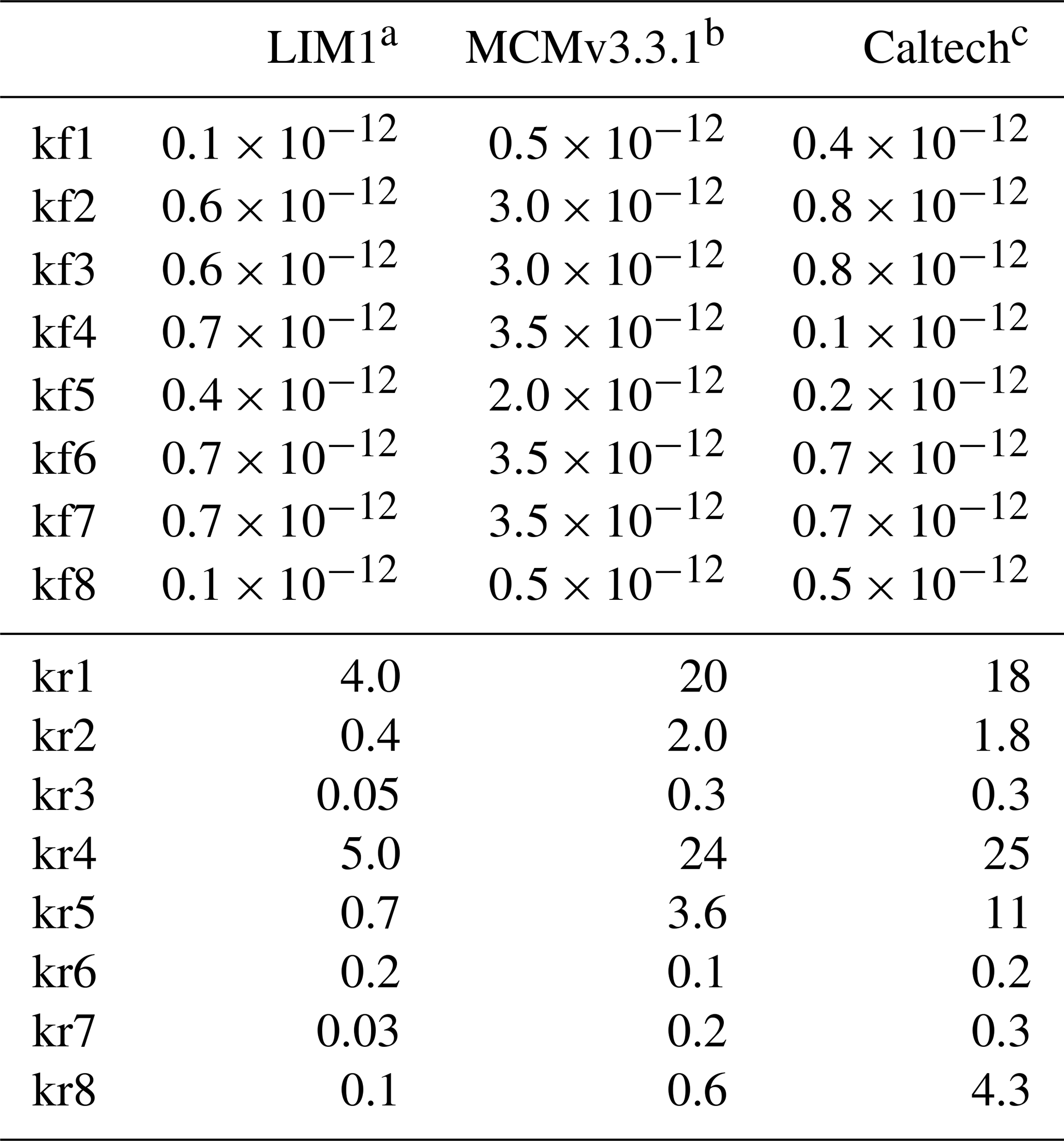
In OH-initiated isoprene oxidation, the first reaction step comprises the formation of six isoprene-RO2 conformers from the addition of the OH radical to the terminal carbon atoms (C1 and C4, 0.91 total yield; Fig. 1), which are in equilibrium and can quickly inter-convert as first suggested in the Leuven isoprene mechanism (LIM) (Peeters et al., 2009) (Fig. 1). The concentration of the different conformers, which is affected by both their losses (unimolecular decomposition and reaction with NO and the hydroxyl and RO2 radicals) and their re-equilibration, can have a large impact on the OH radical concentration. There are three different sets of reaction rate coefficients currently in use in the literature for equilibrium reactions between the isoprene-RO2 conformers (Table 1), differing in the individual rate coefficients by up to a factor of 35. One set is from theoretical calculations in the LIM1 study (Peeters et al., 2014). A second set is currently in use within the Master Chemical Mechanism version 3.3.1 (MCMv3.3.1; Jenkin et al., 2015), whereby the rate coefficients are as described in LIM1 but all increased by a factor of 5. This change was prompted by preliminary results from Caltech (Crounse et al., 2014) and the review by one of the LIM1 authors (Peeters, 2015). Finally, Wennberg et al. (2018), in their recent review paper on the mechanism of isoprene degradation (Caltech mechanism), applied their experimentally optimized parameters, as reported by Teng et al. (2017) (Table 2).
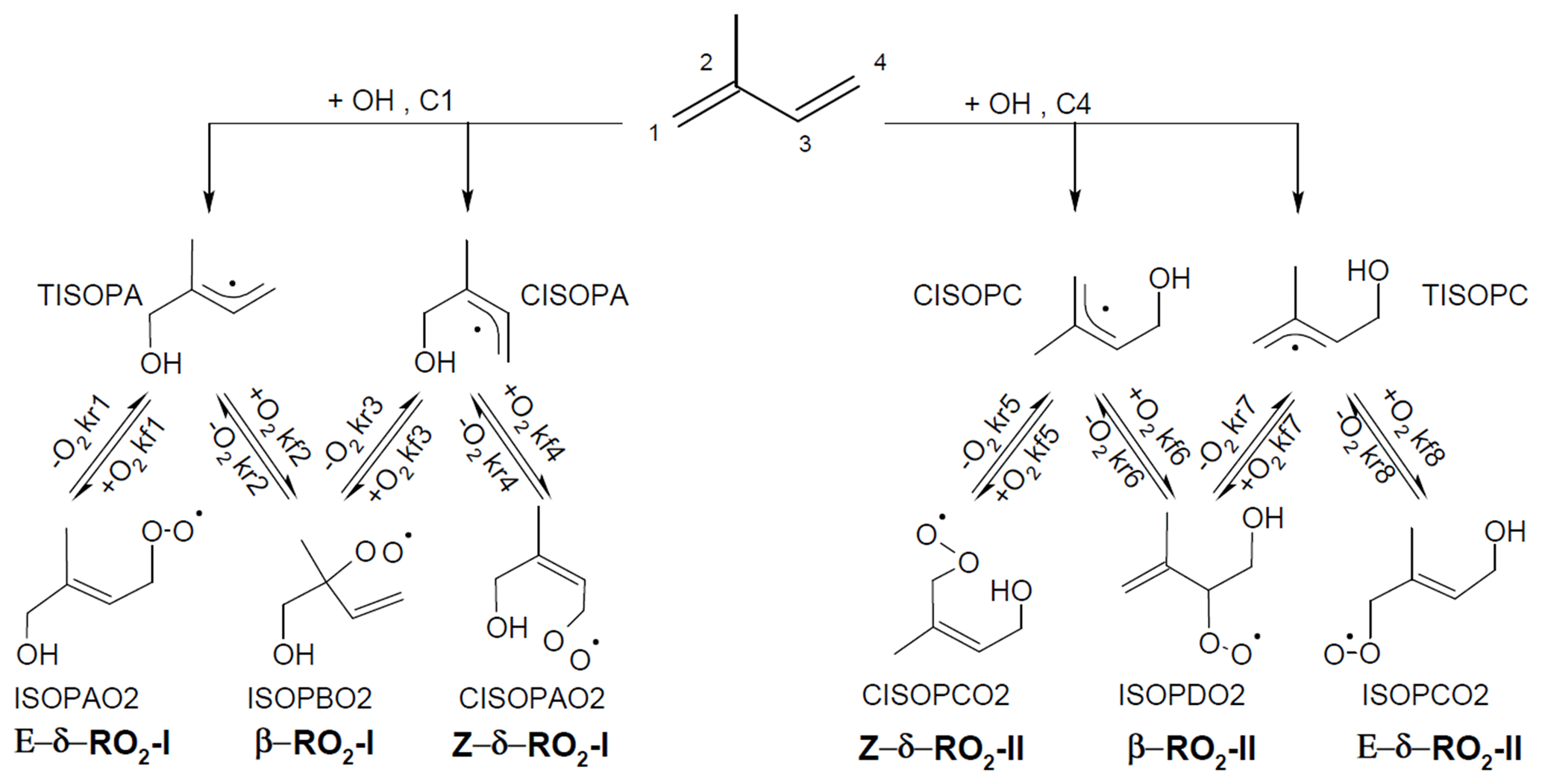
Figure 1Schematic of the equilibrium reactions between OH-isoprene adducts and isoprene-RO2 conformers, as well as their formation reactions. The names used for the different molecules are as in the MCMv3.3.1 (regular text) and in the LIM1 study (bold). The model-specific rate coefficients for each reaction are summarized in Table 1.
Table 1The rate coefficients for the addition of O2 to OH-isoprene adducts and for the re-dissociation of isoprene-RO2 (Fig. 1). The rate coefficients for the oxygen additions (kf; cm3 s−1) are typically temperature-independent or provided at 298.15 K. The rate coefficients for the re-dissociations (kr; s−1) are provided at 298.15 K. The temperature-dependent rate coefficients are given in Table S8.
a Peeters et al. (2014), b Jenkin et al. (2015), c Wennberg et al. (2018).
Four of the six isoprene-RO2 conformers can undergo atmospherically relevant isomerization reactions (Fig. 2). The β-RO2 radicals directly reform OH radicals, together with the oxygenated organic products methacrolein (MACR), methyl vinyl ketone (MVK), and formaldehyde (HCHO), after a 1,5 hydroxy-hydrogen shift (1,5-H shift) (Da Silva et al., 2010) with a slow reaction rate constant ( and s−1 at 298 K for OH addition on C4 and C1, respectively) (Peeters et al., 2014), making this reaction competitive only in the presence of exceptionally low NO levels (<10 pptv).

Figure 2Simplified reaction schematic following OH addition to isoprene on the carbon C1. Only the most relevant reaction paths for OH radical formation are shown; the implemented mechanism includes all six isoprene-RO2 isomers. The schematic illustrates the RO2 unimolecular H shift reactions (1,5-, 1,6-, and aldehyde-H shifts), but all RO2 species undergo competing reactions with NO, HO2, and RO2 radicals (not shown), and the reaction steps shown can represent multiple fast, sequential elementary reactions. The rate coefficients and yields shown are obtained from the theoretical work from this study and from recent experimental and theoretical studies. The same yields for the product of the 1,6-H shift, rate coefficient of the aldehyde-H shift, and photolysis frequencies for HPALD are applied to the chemistry following the OH addition on C4. All rate coefficients are shown for 298 K and a 30∘ solar zenith angle.
The most relevant isomerization reaction, the 1,6 α-hydroxy-hydrogen shift (1,6-H shift), occurs for the Z-δ-RO2 radicals with a fast reaction rate coefficient (measured at 3.6 and 0.4 s−1 at 298 K for OH addition on C4 and C1, respectively, by Teng et al. (2017). These experimental values are used directly within the Caltech mechanism and are in good agreement with the calculated rates in LIM1 (Peeters et al., 2014) (within 40 %). The MCMv3.3.1 is currently using rate coefficients slower by a factor of ∼5 (Table 2). This change was suggested by one of the LIM1 authors (Peeters, 2015) to keep the phenomenological bulk isomerization rate in agreement with previous experimental results on the unsaturated hydroperoxy aldehyde (HAPLD; Fig. 2) formation (Crounse et al., 2011). Following the definition by Peeters et al. (2014), these phenomenological bulk isomerization rates (k(bulk 1,6-H)) are equal to the sum of the isomer-specific 1,6-H shift rate multiplied by its steady-state fraction weighted by their OH addition branching ratio.
Table 2Summary of the relevant differences for assessing the 1,6-H shift impact between available chemical models. All model versions within this work are based on the MCMv3.3.1 (accounting for chamber properties) with only specific rates and yields included in the table adapted from different studies. They also include the follow-up chemistry of di-HPCARP-RO2 as theoretically investigated within this study.
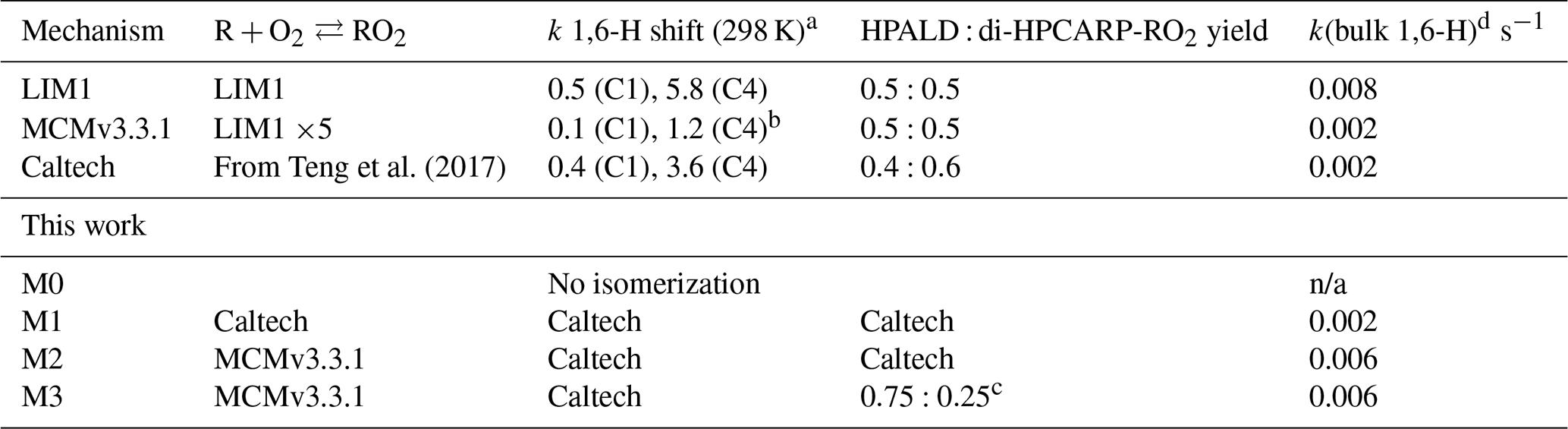
a s−1. b The same rate coefficient as in LIM1 reduced by a factor of 5. c Adapted from Berndt et al. (2019). d For the experimental concentration as observed in Fig. 4. n/a – not applicable
One of the predicted (Peeters et al., 2009, 2014) and measured (Crounse et al., 2011; Berndt, 2012; Teng et al., 2017; Berndt et al., 2019) products following the 1,6-H shift is HPALD, which can photolyse, producing OH radicals (Peeters and Müller, 2010; Wolfe et al., 2012; Liu et al., 2017; Müller et al., 2019) (atmospheric lifetime of ∼1 h). In addition, the formation of a di-hydroperoxy carbonyl peroxy radical (di-HPCARP-RO2, Fig. 2) was predicted by theoretical calculations (Peeters et al., 2014) and inferred in a recent experimental study (Teng et al., 2017). Its fate is a fast unimolecular decomposition (∼1 s−1) with the formation of an OH radical (Wang et al., 2018; Møller et al., 2019) and suggested subsequent elimination of CO, as well as the formation of a di-hydroperoxy carbonyl compound (Peeters et al., 2014; Møller et al., 2019). Large uncertainties remain on the yield of these two products. One earlier experimental study proposes a yield for HPALD of the order of 0.04 with a factor of 2 uncertainty (Berndt, 2012). A more recent experimental study (Teng et al., 2017) suggests a total yield of HPALD of 0.4, distinguishing between β- (0.25) and δ-HPALD (0.15) with large uncertainties in the assignment of the latter; the remainder, 0.6, is assigned to di-HPCARP-RO2 LIM0 (Peeters and Müller, 2010) and LIM1 (Peeters et al., 2014); both proposed a yield of ∼0.5 for HPALD and di-HPCARP-RO2. In contrast, a recent experimental study by Berndt et al. (2019) sets a lower limit of 0.75, with a recent addition (Müller et al., 2019) to the theoretical work within the LIM1 also rationalizing a much higher yield of HPALD (0.74) than previously reported in LIM1. In addition to the above products, both experimental (Berndt et al., 2019) and theoretical (Müller et al., 2019) studies suggest the formation of an hydroperoxy-epoxy-carbonyl compound (∼0.15).
Both currently available explicit isoprene oxidation mechanisms, i.e. the Master Chemical Mechanism (MCMv3.3.1) (Jenkin et al., 2015) and the Caltech mechanism (Wennberg et al., 2018), use a yield of 0.5 and 0.4, respectively, for HPALD, with the Caltech mechanism distinguishing between β- (0.15) and δ-HPALD (0.25). In both models, the only other product formed from the 1,6-H shift is di-HPCARP-RO2.
To summarize, recent experimental and theoretical studies agree that the most relevant isomerization reaction of isoprene peroxy radicals is the 1,6-H shift of Z-δ-RO2. The following kinetic aspects control the impact of the 1,6-H shift of Z-δ-RO2 on the regeneration of OH radicals and the production of oxygenated products:
-
the equilibrium between the isoprene-RO2 conformers, which determines the fraction of Z-δ-RO2 radicals that can undergo fast 1,6-H shift isomerization;
-
the temperature-dependent rate coefficient for the 1,6-H shift itself;
-
the relative yields of HPALD and di-HPCARP-RO2 formed following the 1,6-H shift; and
-
the follow-up chemistry of HPALD and di-HPCARP-RO2.
Despite intensive research as detailed above, there are significant differences between current chemical mechanisms (Table 2); i.e. different sets of rate coefficients are used for the equilibrium reactions, rate coefficients for the 1,6-H shift differ by up to a factor of 5, and the measured yield of HPALD ranges from 0.4 to 0.75.
In this work, new chamber experiments have been performed to test our understanding of the photo-oxidation of isoprene. The experiments are used to test the ability of the explicit mechanisms in MCMv3.3.1 to predict OH radical regeneration from isoprene oxidation over a wide range of NO concentrations (0.15 to 2 ppbv). The chemistry of di-HPCARP-RO2 has been investigated (Novelli et al., 2018a) with high levels of theory, in particular to confirm the role of these radicals in OH radical formation. Model sensitivity studies are applied to identify the isoprene-RO2 conformer equilibrium constants, the 1,6-H shift rate constant, and the HPALD∕di-HPCARP-RO2 branching ratio that provide the best description of the observed radical and trace gas concentrations. The global impact of the optimized isoprene mechanism on the OH radical concentration is shown.
2.1 Quantum chemical and theoretical kinetic calculations
The reactants, transition states, and products in the studied mechanistic branches of the isoprene chemistry were characterized at the M06-2X and CCSD(T) levels of theory. The conformer space for each of these structures was characterized at the M06-2X∕cc-pVDZ level of theory (Dunning, 1989; Zhao and Truhlar, 2008; Alecu et al., 2010; Bao et al., 2017), locating ∼24 000 distinguishable structures from ∼60 000 systematically generated starting geometries. The most relevant conformers (∼850 structures across all reactions examined) were then fully re-optimized at the M06-2X∕aug-cc-pVTZ level of theory (Dunning, 1989). The number of conformers re-optimized at this higher level of theory differs per structure (see Table S1 in the Supplement), but enough were included to cover over ∼80 % of the thermal population at 300 K. Intrinsic reaction coordinate (IRC) calculations were performed on the lowest transition state (TS) to verify the nature of the transition state and to provide the energies used for Eckart tunnelling corrections. Finally, single-point energy calculations at the CCSD(T)∕aug-cc-pVTZ level of theory (Purvis and Bartlett, 1982) were performed on the energetically lowest-lying geometries of each structure to further refine the energy barrier estimates. The thermal rate coefficients were then obtained using multi-conformer transition state theory (MC-TST) incorporating the energetic and rovibrational characteristics of all conformers (Vereecken and Peeters, 2003). Temperature-dependent rate coefficients are derived for the temperature range between 200 and 400 K, and both isomer-specific and bulk phenomenological rate coefficients are provided; the latter incorporate the effect of fast H scrambling in the hydroperoxide-peroxy radical isomers. See the Supplement for a more detailed description of the methodologies involved.
2.2 Atmospheric simulation chamber SAPHIR and experimental procedure
The experiments were conducted in the atmospheric simulation chamber SAPHIR at Forschungszentrum Jülich, Germany. The SAPHIR chamber is designed for the investigation of oxidation processes under atmospheric conditions in a controlled environment. SAPHIR is made of a double-wall Teflon (FEP) film that is inert, has a high transmittance for solar radiation (Bohn and Zilken, 2005), and is equipped with a shutter system that is opened during photolysis experiments, allowing solar radiation to penetrate the chamber. The synthetic air provided to the chamber is mixed from ultra-pure nitrogen and oxygen (Linde, >99.99990 %). Two fans in the chamber ensure complete mixing of trace gases within 2 min. The pressure in the chamber is slightly higher than ambient (∼30 Pa) to avoid external air penetrating the chamber. Due to small leakages and air consumption by instruments, trace gases are diluted at a rate of ∼6 % h−1 due to the replenishment flow. More details regarding the chamber can be found elsewhere (Rohrer et al., 2005; Poppe et al., 2007; Schlosser et al., 2007).
The chamber was cleaned before the experiments by exchanging the chamber air 8 to 10 times with pure synthetic air. Evaporated Milli-Q® water was then introduced into the dark chamber by a carrier flow of synthetic air until a concentration of cm−3 of water vapour was reached. In order to keep the concentration of NO as small as possible after the opening of the shutters, ozone produced by a silent discharge ozonizer (O3onia) was added in the chamber to reach ozone mixing ratios up to 100 ppbv. For experiments at higher concentrations of NO, NO was injected from a gas mixture (Linde, 500 ppm NO in N2) into the chamber by a mass flow controller. After opening the shutter system of the chamber, nitrous acid (HONO) was photochemically formed on the Teflon surface and released into the chamber (Rohrer et al., 2005), and its subsequent photolysis produced OH radicals and NO. Afterwards, isoprene was injected three times at intervals of about 2 h directly from the liquid (99 % purity, Sigma Aldrich). The aim was to reach ∼6 ppbv of isoprene in the chamber after each injection (which corresponds to an OH reactivity between 12 and 15 s−1). Experiments were designed such that chamber-specific sinks (dilution and wall loss of trace gases), and sources of trace gases that are formed in the sunlit chamber, except for nitrous acid, did not influence the results.
Table 3Stereospecific rate coefficients (s−1) at 300 K for the relevant reactions of di-HPCARP-RO2-I. The temperature dependence is given as a Kooij expression, , for the temperature range 200–400 K. The effective bulk rates of reactions are also given, accounting for hydroperoxide H-atom scrambling and aldehyde H migration across all channels. The bottom expression averages the stereospecific rate coefficients for use in simplified models.

* Average of the data for (2R,3R) and (2R,3S) conformers.
2.3 Instrumentation
Table 4 summarizes the instruments available during the experiment, giving time resolution, accuracy, and precision for each instrument. The concentrations of OH, HO2, and RO2 radicals were measured with the laser-induced fluorescence (LIF) instrument permanently in use at the SAPHIR chamber and described previously (Holland et al., 2003; Fuchs et al., 2011). Several studies have been recently published showing the presence of an interference in OH radical detection with the LIF for ambient measurements in some environments (Mao et al., 2012; Novelli et al., 2014; Rickly and Stevens, 2018). The interference depends on the chemical conditions of the sampled air and on the geometry of the different instruments. A laboratory study performed with this LIF instrument (Fuchs et al., 2016) showed only interferences for high ozone concentrations (300–900 ppbv) together with biogenic volatile organic compound (BVOC) concentrations up to 450 ppbv, which are far beyond any condition encountered in this study. Therefore, the OH radical concentration measured by the LIF instrument in this study is considered free from interferences. In addition, OH was measured by differential optical absorption spectroscopy (DOAS) (Dorn et al., 1995) for some of the experiments shown within this study. Numerous inter-comparisons between the LIF and the DOAS instrument in the SAPHIR chamber (Schlosser et al., 2007, 2009; Fuchs et al., 2012) showed very good agreement between these two instruments, giving high reliability to the OH radical measurements performed in the chamber. For the experiments within this study, a slope of 1.1 for the scatter plot of DOAS OH vs. LIF OH was obtained, with a correlation coefficient, R2, of 0.94.
Table 4Instrumentation for radical and trace gas quantification during the oxidation experiment.
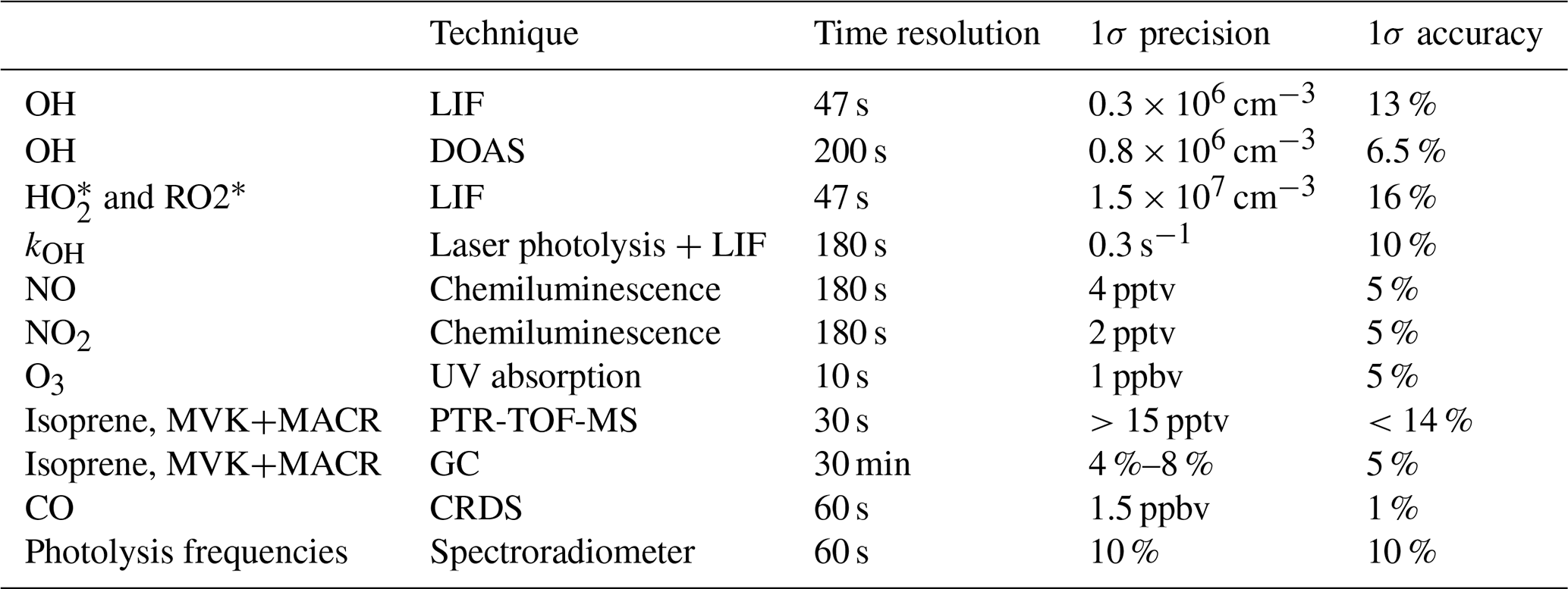
Several studies have proven that RO2 radicals can cause an interference signal in the HO2 radicals measured by conversion to OH after reaction with an excess of NO (Fuchs et al., 2011; Hornbrook et al., 2011; Whalley et al., 2013; Lew et al., 2018). It was shown that a reasonable approach for avoiding the interference is to lower the concentration of NO reacting with the sampled air inside the instrument. During this study, the NO concentration used was low ( cm−3) to minimize the possibility of an interference as described in Fuchs et al. (2011). Still, as the RO2 radicals which originate from the oxidation of isoprene by OH radicals are those able to quickly convert, despite the low NO used, interference from RO2 radicals was still observed. Tests performed on the LIF instrument used for this study showed that, for the conditions the instrument was used in during the experiments, an interference of ∼30 % was observed for isoprene-RO2. As such, the HO2 radical measurement was defined as to indicate the presence of interference from RO2 radicals. As the measured RO2 concentration is obtained from the difference between measured ROx (OH+HO2+RO2) and HO2 radicals, the obtained RO2 radicals are also underestimated due to the interference observed in the HO2 measurement and will be marked as . The OH reactivity (kOH), the inverse lifetime of OH, was measured by a pump-and-probe technique coupled with a time-resolved detection of OH by LIF (Lou et al., 2010; Fuchs et al., 2017). Isoprene and the sum of MVK and MACR were measured by a proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS; Lindinger et al., 1998; Jordan et al., 2009) and a gas chromatography system (GC; Wegener et al., 2007). As the PTR-TOF-MS and the GC were calibrated only for the species listed in Table 4, concentrations for other species were not available. Carbon monoxide (CO), carbon dioxide (CO2), methane (CH4), and water vapour were measured by an instrument applying cavity ring-down spectroscopy (CRDS; Picarro). NO and nitrogen dioxide (NO2) were measured by chemiluminescence (CL; Ridley et al., 1992) and O3 by UV absorption (Ansyco). Photolysis frequencies were calculated from measurements of solar actinic radiation by a spectroradiometer (Bohn et al., 2005; Bohn and Zilken, 2005).
2.4 Model calculations
The measured radicals and trace gases were modelled with a zero-dimensional box model using chemical mechanistic information from the Master Chemical Mechanism downloaded via the following website: http://mcm.leeds.ac.uk/MCM (last access: November 2019). The MCMv3.3.1 was released in 2015 with newly updated isoprene chemistry in line with LIM1 chemistry, updated and/or optimized to recent experimental results, as described in Jenkin et al. (2015). The most relevant changes for this study are the inclusion of the equilibrium reactions between the OH-isoprene adducts and isoprene-RO2 (Fig. 1) and the inclusion of isomerization reactions for isoprene-RO2 radicals (Table 2). Further, the OH addition to central carbon atoms (C2 and C3, Fig. 1) and following chemistry was implemented.
Several chamber-specific properties were implemented in the model. First, a dilution rate was applied to all the trace gases present in the model to account for dilution from the replenishing flow of the chamber. The background production of HONO and HCHO known to occur in the sunlit chamber (Rohrer et al., 2005; Karl et al., 2006) was parameterized by an empirical function that depends on temperature, relative humidity, and solar radiation. For the experiments shown in this study, the background OH reactivity in the chamber was at most 1 s−1 and was parameterized with a co-reactant Y added to the model, which converts OH to HO2 in the same way as CO does (Fuchs et al., 2012, 2014; Kaminski et al., 2017). The concentration of Y was adjusted to match the observed OH reactivity during the zero-air phase of the experiment and was kept constant throughout the experiment. As shown in a previous study (Novelli et al., 2018b), the unknown chemical nature of the background reactivity that dominates the loss of OH radicals for the zero-air phase of the experiment has a negligible impact once the main reactant, in this case isoprene, is added, with total OH reactivity as high as 20 s−1.
Photolysis frequencies for O3, NO2, HONO, hydrogen peroxide (H2O2), formaldehyde (HCHO), acetone, glyoxal, MVK, and MACR were constrained to values calculated from the measured actinic flux. For HPALD and peroxy acid aldehyde (PACALD), photolysis frequencies of MACR scaled up by a factor of 100 and 2, respectively, were used (Fuchs et al., 2013). All the other photolysis frequencies present in the model were first calculated for clear-sky conditions according to the MCMv3.3.1 and then scaled by the ratio of measured to calculated j(NO2) to account for clouds and transmission of the chamber film. The model was constrained to measured chamber pressure (ambient pressure) and temperature, as well as water vapour, NO, NO2, and O3 concentrations. Model values were re-initiated at 1 min intervals. Isoprene injections were implemented in the model by applying an isoprene source only active at the time of the injection, adjusted in strength to reproduce the observed change in OH reactivity at the injection time. The modelled OH reactivity used for comparison against the measurement is the total modelled OH reactivity excluding the reactivity of isoprene hydroxy hydroperoxides (ISOPOOHs) (Fig. 2), as for these compounds the OH radicals are recycled at a timescale much shorter than the OH lifetime observed in the kOH instrument, negating their measurable OH reactivity. Measurements of MVK and MACR by PTRMS and GC are affected by interferences from ISOPOOHs (Rivera-Rios et al., 2014). For this reason the measured data are compared with the sum of MVK and MACR together with ISOPOOHs (all isomers); the same sensitivity for MVK, MACR, and ISOPOOHs is assumed. Due to RO2 interference in the HO2 measurement, modelled HO2 concentrations increased by a small fraction of modelled RO2 (30 % of RO2 radicals from isoprene and 10 % of RO2 radicals from MVK and MACR; Fuchs et al., 2013). Likewise, the measured values are compared against the difference between modelled ROx and , rather than the uncorrected RO2 concentrations, to account for this interference.
The chemistry of di-HPCARP-RO2-I and di-HPCARP-RO2-II, originating from the addition of the OH radical on C1 and C4, respectively, is implemented in the model based on our explicit study of the di-HPCARP-RO2-I reactions, and the chemistry for di-HPCARP-RO2-II could thus need refining in future work (see also below).
This model (MCMv3.3.1, Table 2) served as the basis of all model calculations done in this work, with variations as defined below. Table 2 summarizes the additional model runs performed.
-
M0 was constructed by removing all isomerization reactions (no isomerization, Table 2) from the MCMv3.3.1 model (see the Supplement and Table S2).
-
M1 (Caltech, Table 2) was built by using the rate coefficients for the reversible addition of O2 to OH-isoprene adducts, the rate coefficient for the 1,6-H shift of Z-δ-RO2 radicals, and the relative yield of HPALD∕di-HPCARP-RO2, as applied in the Caltech mechanism (Table S3).
-
M2 is the same as M1 but using the rate coefficients for the reversible addition reactions of O2 to OH-isoprene adducts as applied in the MCMv3.3.1 (Tables 2 and S4).
-
M3 is identical to M2 but using a relative yield of HPALD∕di-HPCARP-RO2 of 0.75:0.25 adapted from Berndt et al. (2019) (Tables 2 and S5).
Within this study only δ-HPALD (called HPALD) and its following chemistry are included in the different models. For M1 and M2 the sum of the δ- and β-HPALD yield is used as HPALD (Table S4). The identification of β-HPALD and its following chemistry is uncertain, but within the Caltech mechanism it will form the same products as formed from δ-HPALD, albeit with large uncertainties on the yields and rate coefficients (Wennberg et al., 2018) and without an experimentally accessible way to distinguish between the two within this study. Following the same reasoning, within the M3 model, we do not include the hydroperoxy-epoxy-carbonyl compound; as its chemistry cannot be univocally probed, its yield is comparatively small (∼15 %, see above), and it has little influence on the topics investigated in this work.
2.5 Global model
The ECHAM–MESSy Atmospheric Chemistry (EMAC) model is a numerical chemistry and climate simulation system that includes submodels describing tropospheric and middle atmosphere processes and their interaction with oceans, land, and human influences (Jöckel et al., 2010). It uses the second version of the Modular Earth Submodel System (MESSy2) to link multi-institutional computer codes. The core atmospheric model is the fifth-generation European Centre Hamburg general circulation model (ECHAM5) (Roeckner et al., 2006). For the present study we applied EMAC (ECHAM5 version 5.3.02, MESSy version 2.53.0) in the T106L31ECMWF resolution, i.e. with a spherical truncation of T106 (corresponding to a quadratic Gaussian grid of approximately 1.1 by 1.1∘ in latitude and longitude) with 31 vertical hybrid pressure levels up to 10 hPa. The applied model setup comprised the submodel MECCA (Module Efficiently Calculating the Chemistry of the Atmosphere) to calculate atmospheric chemistry using the Mainz Organic Mechanism (MOM) (Sander et al., 2011). The mechanism was adapted to the changes proposed in this study (Table S7). In addition, the submodel MEGAN (Model of Emissions of Gases and Aerosols from Nature) was used to simulate biogenic emissions of tracers, including isoprene (Guenther et al., 2006). Global isoprene emissions were scaled to 595 Tg yr−1, which is the best estimate by Sindelarova et al. (2014). The model was run for 1.5 years (summer 2011–2012), during which the first half-year was used as spin-up and 2012 was used for analysis.
3.1 Kinetics of the di-HPCARP-RO2 H-migration reactions
The di-HPCARP-RO2 chemistry was studied in more detail compared to earlier published work (Wang et al., 2018; Møller et al., 2019), with more reliable kinetic methodologies, to obtain the isomer-specific rate coefficients, the phenomenological (bulk) rate coefficients, and the subsequent chemistry. Table 3 lists the rate coefficients at 300 K and the Kooij expressions for the key reactions in the di-HPCARP-RO2-I reaction system for the temperature range 200–400 K. As expected from the higher energy barriers and entropic considerations, the migration of non-aldehyde H atoms is not competitive, nor is HO2 elimination. The fastest reactions are, for all di-HPCARP-RO2-I intermediates, the hydroperoxide-H scrambling across all peroxyl sites. This result is anticipated, as fast scrambling has been known for several years (Miyoshi, 2011; Jorgensen et al., 2016; Knap and Jorgensen, 2017; Møller et al., 2019; Praske et al., 2019). Given that these reactions outrun the next fastest reaction by over an order of magnitude, it can be assumed that the different di-HPCARP-RO2-I isomers are in steady-state equilibrium and can be considered a unified pool of reactants in atmospheric models for the purpose of unimolecular reactivity. The 1,4-aldehyde-H-migration reaction is comparatively slow owing to its higher energy barrier (see Table S1, Fig. 3), and the formation of the tri-hydroperoxide acyl radical tri-HPACYL will thus occur mostly by 1,5- and 1,6-H migration in the di-HPCARP-RO2-I reactant pool. Table 3 lists the rate coefficients of the elementary processes, but for atmospheric modelling the more relevant numbers are the bulk k(T) expressions that account for the H scrambling and the combined flux across the 1,4-, 1,5-, and 1,6-H migrations listed at the bottom of the table. The stereospecific rate coefficients are not all that different and can be expressed within a factor of 2 to 3 as a single Kooij expression across the temperature range 200–400 K as follows (see also Fig. S1 in the Supplement):
The effective rate of acyl radical formation by aldehyde-H migration, i.e. accounting for the rapid re-equilibration across the di-HPCARP-RO2-I isomers and the different H shift channels, is then of the order of 10 s−1 at 300 K. For the (2R, 3R) and (2S, 3S) stereoisomers, 63 % of the reaction flux at 300 K passes through the 1,5-aldehyde-H migration, with 35 % undergoing a 1,6-H migration. For the (2R, 3S) and (2S, 3R) isomers, 1,6-aldehyde-H migration constitutes over 99 % of the acyl radical formation. The estimated uncertainty on the rate coefficient is about a factor of 5, mostly due to the current use of non-conformer-specific tunnelling.

Figure 3Potential energy surface for the aldehyde-H shift reaction showing the multiple competing reactions. A set of fast H-migration reactions ultimately leads to the formation of a tri-hydroperoxy acyl radical, tri-HPACYL. The main fate of this radical is shown by explicit theoretical calculations to be CO elimination, forming DHP-MVK; O2 addition to a tri-hydroperoxy acylperoxy radical has only a minor contribution.
3.2 Elimination of CO from tri-hydroperoxy acyl radicals
It has been assumed in most models in the literature (Peeters et al., 2009, 2014; Jenkin et al., 2015; Wennberg et al., 2018) that the tri-hydroperoxy acyl radical formed, tri-HPACYL, will eliminate CO, followed by OH elimination and the formation of a di-hydroperoxy carbonyl compound, DHP-MVK (Fig. 3). All these models seem to be based ultimately on an estimate by Peeters et al. (2014), who predicted that the reaction would proceed via a 1,4-H shift, forming tri-HPACYL intermediates with an internal energy of the order of 22–25 kcal mol−1, rapidly losing CO with a barrier ≤7 kcal mol−1. This work, however, shows that these predicates do not represent the chemistry accurately, and the fate of tri-HPACYL must be examined in more detail.
The lower energy barriers of the effective aldehyde-H-migration processes imply that the acyl radical tri-HPACYL is formed with a significantly lower energy content, 17–19 kcal mol−1, than estimated by Peeters et al. (2014), reducing the likelihood of chemically activated decomposition. Note that the H-migration reactions are typically found to be in the high-pressure limit (Miyoshi, 2012; Peeters et al., 2014; Xing et al., 2018; Møller et al., 2019), and multi-step chemical activation does not contribute significantly. Furthermore, we found that the CO elimination barrier in tri-HPACYL-I, ∼8 kcal mol−1 (see Table S1), is higher than estimated by Peeters et al. (2014) (≤7 kcal mol−1), further hampering prompt decomposition. With only ∼10 kcal mol−1 excess internal energy, as opposed to 15–18 kcal mol−1 as proposed by Peeters et al. (2014), a significant fraction of the nascent acyl radical tri-HPACYL could be thermalized, and the resulting slower decomposition process could potentially allow for O2 addition on the acyl radical site. However, based on MC-TST calculations incorporating all conformers, we found that thermal decomposition of the tri-HPACYL-I acyl radicals is still sufficiently fast to dominate over O2 addition; i.e. even when assuming a Boltzmann energy distribution, the predicted rate coefficient of s−1 at 300 K (see Table 3) is significantly higher than the effective O2 addition rate for acyl radicals, experimentally measured at s−1 in atmospheric conditions (Sehested et al., 1998; Blitz et al., 2002; Park et al., 2004; Baulch et al., 2005; Atkinson et al., 2006; Carr et al., 2011). One could counter that the presence of –OOH groups might stabilize the acylperoxy radicals formed in the O2 addition (e.g. by H bonding), thus increasing the addition rate coefficient above those reported for the smaller acyl radicals in the literature. Sample calculations on a smaller proxy with substituted acetyl radicals (see Table S1) revealed no evidence that a hydroperoxide group interacts with the oxygen atom moiety in a way that reduces the entrance energy barrier (thus increasing the capture rate coefficient) or stabilizes the adduct (thus reducing re-dissociation). From these results, we conclude that CO elimination will be the dominant fate at 300 K for the tri-HPACYL-I acyl radicals formed in aldehyde-H migration, with O2 addition as a minor channel. Whether CO elimination occurs promptly or in a thermal reaction is then a moot issue.
Based on a preliminary version of our results (Novelli et al., 2018a), Peeters and coworkers (Müller et al., 2019) now suggest that tri-HPACYL-II acyl radicals (differing from the case I acyl radical by the position of the methyl group) would not eliminate CO due to a higher CO elimination energy barrier (Méreau et al., 2001) compared to the tri-HPACYL-I acyl radicals we explicitly characterized above. We have as yet been unable to dedicate the required significant computational resources for an explicit study of di-HPCARP-RO2-II and tri-HPACYL-II, so this issue cannot be resolved at this time. We can, however, estimate the tri-HPACYL-II thermal CO elimination rate by assuming that case I and case II reactions are entropically similar and increasing the barrier for tri-HPACYL-II by the difference between a tertiary (case I) and secondary (case II) product radical as calculated by Méreau et al. (2001). The obtained rate coefficients at 298 K ( s−1 for the (R, R) and (S, S) stereoisomers; s−1 for the (R, S) and (S, R) stereoisomers) remain competitive against O2 addition. Though these channels appear no longer truly dominant thermally, CO elimination from tri-HPACYL-II can clearly not be discounted with any degree of confidence based on such estimates, especially as any chemical activation afforded by the preceding H migration would further shift the subtle competition towards higher CO yields. Quantifying this yield theoretically would be a very committed effort, as it requires explicit calculation of the full conformation space, with chemical activation depending on the energy-specific state density, conformer-specific tunnelling, and collisional energy transfer across the thousands of di-HPCARP-RO2-II and tri-HPACYL-II conformers; this is beyond the scope of the current paper. Therefore, to keep the model simple and for lack of better information, the same aldehyde-H shift is implemented for both HPCARP-RO2 isomers, both followed by the formation of CO, OH radicals, and DHP compounds.
3.3 Comparison to literature theoretical work
Two earlier studies examined the di-HPCARP-RO2 chemistry theoretically. A first study by Wang et al. (2018) was based on a partial characterization of the conformational space. While some of the provided rate coefficients are comparable to our values, these predictions carry a larger uncertainty due to the limited number of conformers examined, and differences of over an order of magnitude are found compared to our predictions, leading to qualitative changes in the predicted fate of the intermediates. The methodology of the second study, by Møller et al. (2019), is more comparable to that used in our work, with the main difference being the methodology used to screen the conformational space and select the conformers included in the rate calculations. As discussed in more detail in the Supplement, we find that this study only incorporates half or less of the population in the kinetic predictions, relying on the cancellation of error to a far greater extent than our more rigorous population description. Though for most reactions the differences in the predicted rate coefficient at 298 K are small, we find some values that differ by about an order of magnitude. We surmise that this is mostly due to the impact of the population truncation in the Møller et al. (2019) study compared to the full population used our work (e.g. 11 conformers versus ∼1500 conformers included), making our predictions more robust in this respect. Improved conformer screening methodologies, balancing completeness and accuracy against computational cost, will help to converge the results of the two similar methodologies; this is discussed briefly in the Supplement.
Figure 4 shows the evolution of trace gas concentrations for an experiment with three separate injections of isoprene and for which the NO concentration was below 0.15 ppbv for the entire duration of the experiment. For all three isoprene injections it is possible to observe large discrepancies between measured trace gases and model calculations when using the MCMv3.3.1 or the M1 model. Both models underestimate the measured OH radical concentration with a ratio of measured to model data of 0.7±0.07 and overestimate the measured sum or MVK, MACR, and hydroxyl hydroperoxides (ISOPOOHs; Fig. 2) by almost a factor of 2. The similarity between the two models, despite M1 including a factor of 3 faster 1,6-H shift, is due to the different distribution of the isoprene-RO2 conformers. Specifically, a much smaller fraction of Z-δ-RO2 radicals for M1, ∼0.004, is formed compared with 0.015 for the MCMv3.3.1 model, thus reducing the contribution of the 1,6-H shift. Although a larger fraction of Z-δ-RO2 radicals are formed within the MCMv3.3.1 model, the slow 1,6-H shift used also results in an underestimation of the OH radical concentrations. For both models, for conditions under which the NO concentration is lower than 0.2 ppbv, i.e. under which isomerization reactions should become more relevant, ∼30 % of the total loss of isoprene-RO2 conformers (weighted by their abundance) occurs via isomerization reactions. Compared with a simulation without isomerization reactions (M0) there is an improvement in the reproduction of the measured trace gases, but still the importance of the isomerization reactions is underestimated. The best agreement between measured and modelled trace gases was achieved when the equilibrium reactions between the isoprene-RO2 conformers providing a larger fraction of Z-δ-RO2 radicals in combination with the faster 1,6-H shift were used (M2, Fig. 5). Ratios of measured to modelled data of 0.97±0.10 and 0.98±0.07 for the OH radicals and of the sum of the oxidation products MVK, MACR, and ISOPOOHs, respectively, are found. Also, the increase in the carbon monoxide (CO) concentration, of which nearly one-quarter is explained by the CO elimination of di-HPCARP-RO2, is well captured by the model calculations (ratio of measured to modelled data of 0.98±0.05). In comparison with MCMv3.3.1 and M1 model runs, ∼50 % of the total loss of isoprene-RO2 conformers proceeds via isomerization reactions. In addition, both the MCMv3.3.1 and M1 models predict a larger concentration of ISOPOOHs compared to the optimized model M2 due to the different distribution of isoprene-RO2 conformers. This will cause a larger expected concentration of new particles formed during the oxidation of isoprene due to the subsequent degradation products of ISOPOOHs, which includes epoxides (St. Clair et al., 2016).
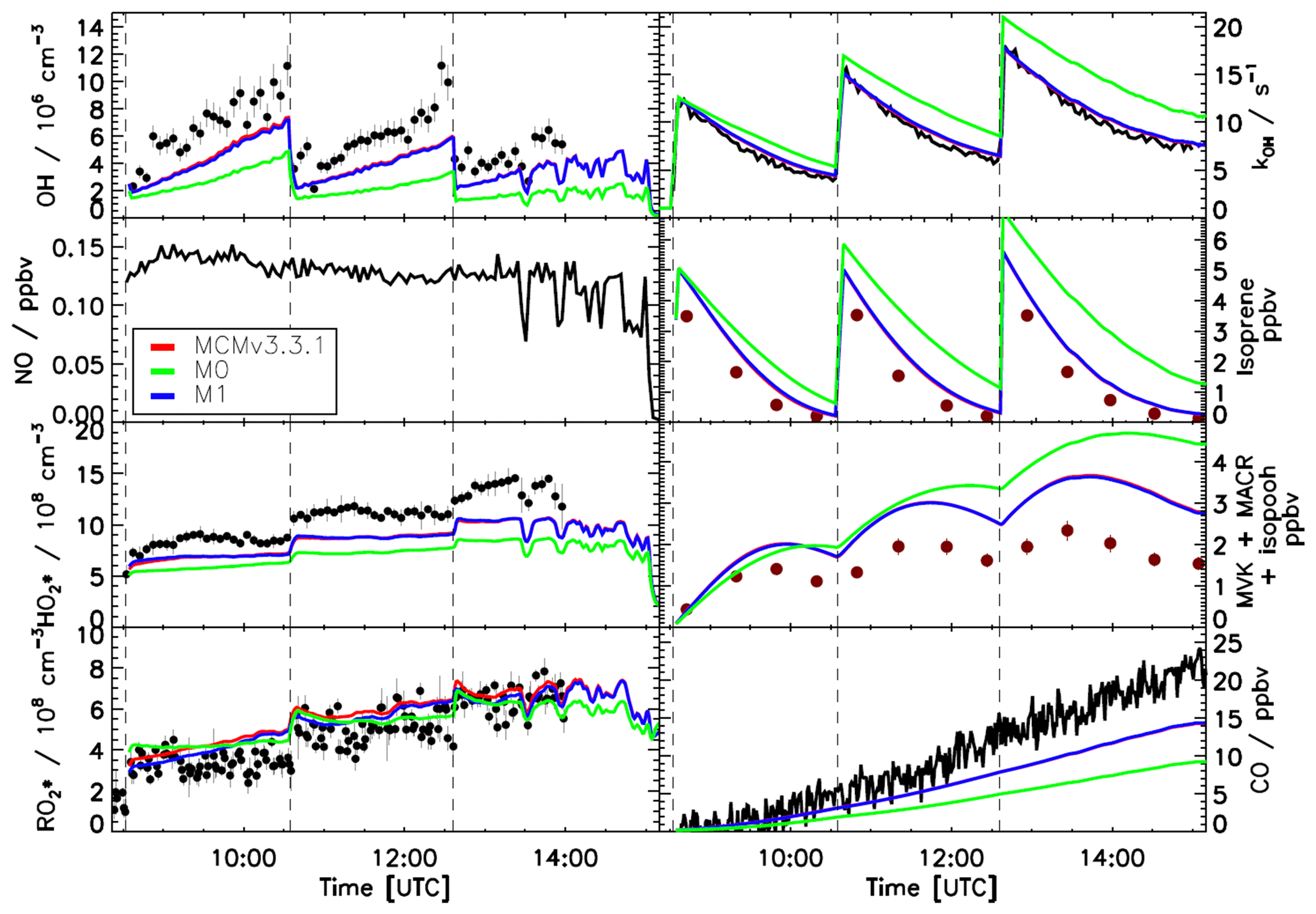
Figure 4Comparison of modelled and measured trace gases for an experiment with NO <0.2 ppbv. Measured time series of radicals and OH reactivity (LIF), isoprene and MVK+MACR+ISOPOOHs (GC), and CO (Picarro) are compared to model calculations. Vertical dashed lines indicate the times when isoprene was injected. No good agreement is observed when using the MCMv3.3.1 or a modified version (M1, Table 2) including isoprene-RO2 conformer equilibrium reactions as included in the Caltech mechanism. Error bars represent 1σ standard deviation.
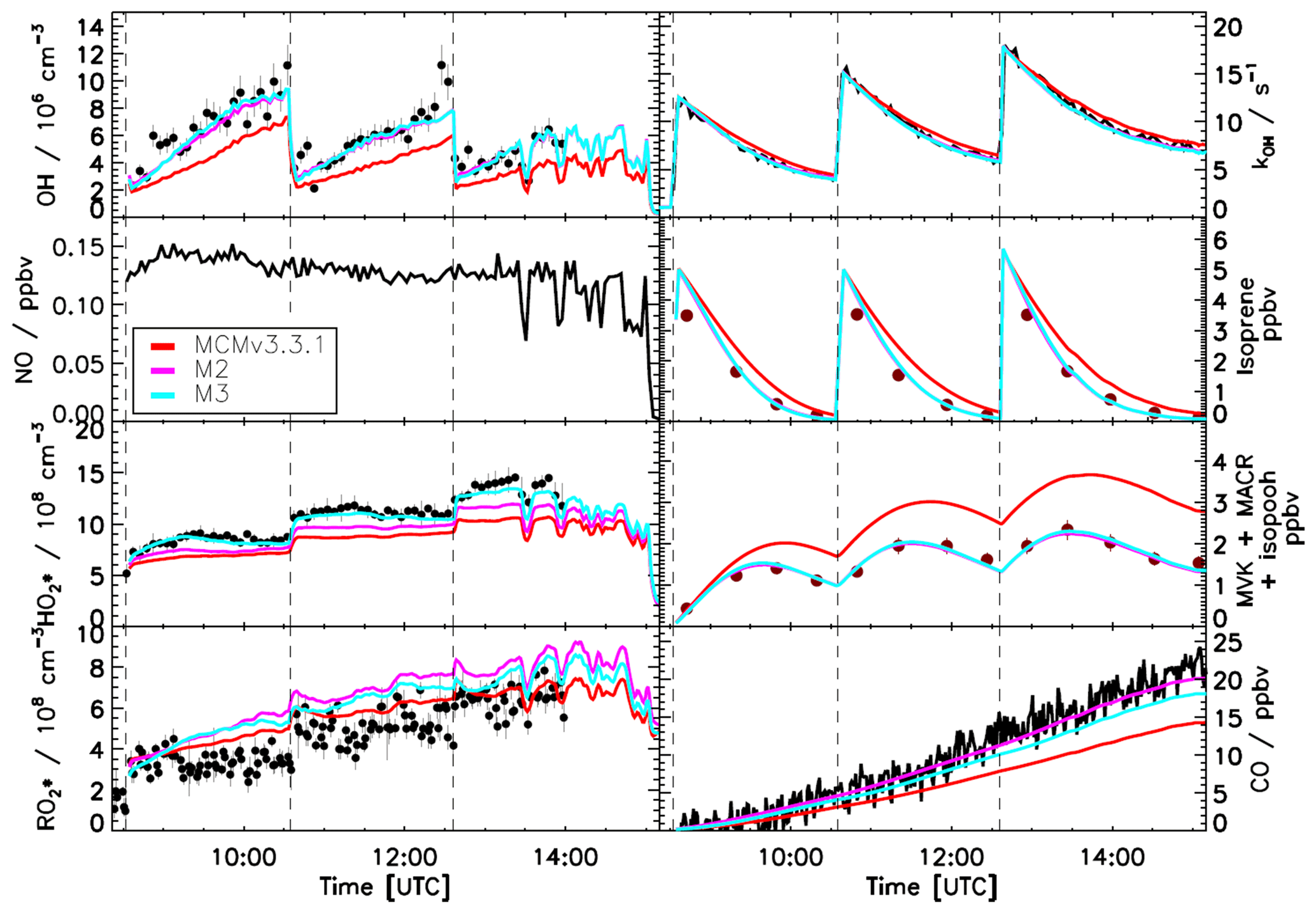
Figure 5Comparison of modelled and measured trace gases for an experiment with NO <0.2 ppbv. Measured time series of radicals and OH reactivity (LIF), isoprene and MVK+MACR+ISOPOOHs (GC), and CO (Picarro) are compared to model calculations. Vertical dashed lines indicate the times when isoprene was injected. Good agreement is observed when using M2 or M3 (Table 2), which use a different yield for HPALD of 0.40 and 0.75, respectively. Within both models, ∼50 % of isoprene-RO2 radicals (weighted by their abundance) are lost via isomerization reactions. Error bars represent 1σ standard deviation.
The yields of the chemical compounds formed following the 1,6-H shift of Z-δ-RO2 radicals carry a large uncertainty as summarized in the Introduction. Two HPALD yields are currently described in the literature: one in the range of 0.4 to 0.5, used within the Caltech mechanism and the MCMv3.3.1, and the other exceeding 0.75 based on the most recent literature. To quantify the impact of the uncertainty on the HPALD yield, a sensitivity model run was performed by changing the yield of HPALD from 0.4 to 0.75, with the yield of di-HPCARP-RO2 set to 0.25 (M3, Table 2). As can be seen in Fig. 5, increasing the yield of HPALD by almost a factor of 2 does not have a large impact on model reproduction of the measured trace gases. The degradation of HPALD and di-HPCARP-RO2 is followed by the formation of OH radicals and CO. On the timescale of the experiments in the chamber, it is not possible to distinguish between the relatively slow formation of OH radicals from the photolysis of HPALD (∼1 h) from their formation from the fast aldehyde-H shift of di-HPCARP-RO2 (∼0.1 s). This is not entirely true for the CO, for which the measurement indicates a faster formation rate, better agreeing with the model and including a larger yield towards di-HPCARP-RO2 (M2 model). A better agreement is, however, observed between measured and modelled radicals for the HPALD 0.75 model, although this remains within the uncertainty of the measurement, due to the formation of HO2 radicals together with HPALD following the 1,6-H shift.
For experiments transitioning from high (1.5 ppbv) to low (0.2 ppbv) NO (Fig. 6) all the models are able to reproduce (within 10 %) the measured trace gases and the OH reactivity for the first injection of isoprene when the NO concentration is above 0.5 ppbv. At this NO level, the OH production is mainly controlled by the reaction between HO2 radicals and NO; therefore, the impact of the isomerization reactions on the OH radical production is marginal. As soon as the concentration of NO decreases below this threshold, larger discrepancies between the model calculations and the measured trace gases can be observed.

Figure 6Comparison of modelled and measured trace gases for an experiment with variable NO concentrations, ppbv. Measured time series of radicals and OH reactivity (LIF), isoprene and MVK+MACR+ISOPOOHs (GC), and CO (Picarro) are compared to model calculations. Vertical dashed lines indicate the times when isoprene was injected. All models are able to reproduce the observed trace gases within their uncertainties for NO >0.2 ppbv.
One additional model run (Fig. S5) was performed by re-implementing the original LIM1 within the MCMv3.3.1 model (Table 2), which includes a factor 5 slower equilibrium reactions between the isoprene-RO2 conformers and a factor of 5 faster 1,6-H shift. Despite the large reduction in the equilibrium reactions between the isoprene-RO2 conformers the LIM1 model run can reproduce the measured data as well as M2 and M3 for all concentrations of NO investigated in this study, as a change of only 5 % in the fraction of Z-δ-RO2 radicals formed is observed.
When comparing the phenomenological bulk isomerization rate among the different models tested within this study calculated for the low NO experiment (Table 2) a similar value is observed for both the MCMv3.3.1 and M1 models. This is to be expected as both models are optimized to reproduce the phenomenological bulk isomerization rate as measured from the formation rate of HPALD (Crounse et al., 2011). In addition, in a study by Jenkin et al. (2019), the MCMv3.3.1 and M1 models are compared for different NO values and show no significant differences, as also observed within this study. On the other hand, the value obtained from this study is in good agreement with the LIM1 theoretical calculations and is needed to bring measurements and model results into agreement. Between these two groups of models, the bulk rate differs by a factor of 3 to 4.
The extensive range of NO concentrations in the experimental studies, reaching up to 2.0 ppbv, allowed for the exploration of the ability of the models to reproduce the measured data and to quantify the efficiency of the regeneration of OH radicals across a wide range of atmospheric conditions (Fig. 7) by drastically changing the competition between the isomerization reactions of RO2 and the RO2 + NO reactions.
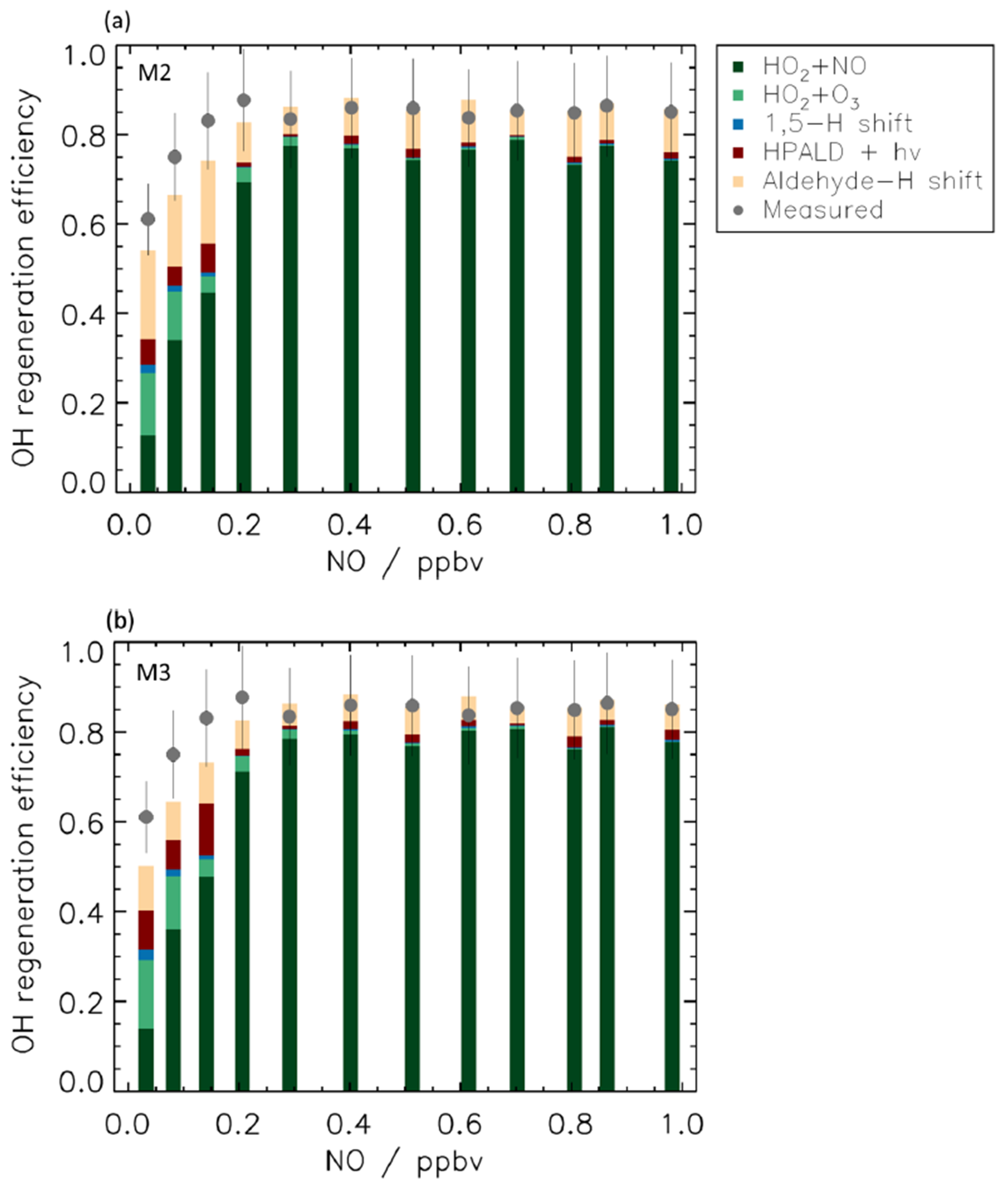
Figure 7OH regeneration efficiency at different NO concentrations. The experimental OH regeneration efficiency (RE) is compared with the modelled one (M2, panel a; M3, panel b) for different NO values. The modelled OH RE is colour-coded by its main contributors (see Table S6 for more details). The RE is sustained at low levels of NO by the contribution from isomerization reactions, in particular by the aldehyde-H shift and its products. For [NO] higher than 0.3 ppbv, most of the OH recycling originates from the reaction of HO2 with NO, but, although to a small extent, isomerization reactions still contribute up to 2 ppbv NO as the 1,6-H shift is still a competitive loss path for Z-δ-RO2 radicals. The relatively large contribution to RE by the reaction between HO2 radicals and O3 at low NO is due to the large concentration of O3 (90 ppbv) needed in the simulation chamber to maintain low values of NO, and it is not representative of ambient conditions. Error bars (1σ) for the measured OH regeneration efficiency include the accuracy of the measurements.
The efficiency of OH regeneration, noted RE henceforward, is defined as the number of OH radicals that are produced after one OH radical has reacted with isoprene. It is calculated as the ratio of the OH regeneration rate R and the OH loss rate L. The modelled R quantifies the OH production via radical chain reactions (HO2+NO, HO2+O3 and isomerization of isoprene-RO2) and the photolysis of HPALD as produced in the isomerization reactions. It also includes the OH regenerated from the direct products of HPALD photolysis and the aldehyde-H shift of di-HPCARP-RO2 and following products as well as the OH regenerated from the aldehyde-H shift of the MACR-RO2 (Table S6). L represents the OH loss by reaction with isoprene and its products. As such, the model values for the OH regeneration efficiency represent a lower limit. The measured RE is obtained from the difference between the total OH loss rate and the primary OH production rate (ozone and nitrous acid photolysis) divided by the total OH loss rate.
Here, most values used ([OH], kOH, [O3], [H2O], jO(1D), jHONO) are experimentally measured quantities. Only the HONO concentration was not measured but taken from the model; y is the fraction of O(1D) reacting with water vapour multiplied by the OH yield of the O(1D) + H2O reaction.
Good agreement is found between measured and modelled OH regeneration efficiency at all values of NO within the uncertainty of the measurements when using either M2 or M3 (Fig. 7), suggesting that all relevant OH production pathways are included.
In environments influenced by anthropogenic emissions, with NO values higher than 0.2 ppbv, 75 % of OH radicals are regenerated by the reaction of HO2 radicals with NO. In contrast, at the lowest NO values representative of rainforest conditions, only 10 % of OH radicals reacting with isoprene are reformed by the HO2 + NO reaction. This decrease in the OH RE from radical reactions with NO is partly compensated for by an increased contribution of regeneration from RO2 isomerization reactions such that the total OH RE is still approximately 0.5 at the lowest NO concentration investigated. Though contributions of isomerization reactions to the OH RE diminish with increasing NO concentrations, their reaction rate coefficients are high enough to still constitute a competitive loss path for Z-δ-RO2 radicals even at 2 ppbv of NO, with 10 % of the OH radicals consumed still regenerated by RO2 isomerization reactions. The differences in the contribution of the isomerization reactions to the OH RE found for [NO] above 0.3 ppbv are mainly due to differences in ambient temperature impacting the isomerization rate coefficient. Although no experiments are available for levels of NO lower than 0.15 ppbv, a model simulation for the OH RE for up to 0.005 ppbv of NO indicates that the value of OH RE remains constant at around 0.5.
Among the isomerization reactions, the 1,5-H shift contributes less than 5 % to OH radical regeneration, with the large majority of the OH radical regenerated by the products following the 1,6-H shift. This is not surprising as the 1,5-H shift rate coefficient is ∼2 orders of magnitude slower than the 1,6-H shift. A more detailed analysis of the contribution of HPALD vs. Di-HPCARP-RO2 and its products to OH regeneration is hindered by the uncertainty on their relative yield. Anyway, it is interesting to see that when a larger yield for di-HPCARP-RO2 (0.6, M2) is applied, the largest fraction of the OH radical is regenerated via the aldehyde-H shift of di-HPCARP-RO2 and its direct di-hydroperoxy carbonyl products (∼70 %; Fig. 7a). When using a yield of HPALD of 0.75 (Fig. 7b) the photolysis of HPALD and the following direct products increase their contribution from 12 % to 60 %. Still, despite a much smaller yield (0.25) compared to HPALD, di-HPCARP-RO2 and direct products contribute up to 30 % to OH radical regeneration due to the fast aldehyde-H shift.
The magnitude of the OH RE observed in this study, however, remains much lower than anticipated from OH concentration measurements in field campaigns under similar conditions, requiring an OH RE of nearly 1 to reproduce the observations (Rohrer et al., 2014). The reason for the discrepancy is still not fully understood. On the one hand, the experiments in the chamber refer only to isoprene chemistry, while the field studies, although performed in areas with large emissions of isoprene, include several different organic compounds which could contribute to the OH concentration. On the other hand, it could be that part of the measured OH radical concentrations in field campaigns was due to an interference. However, an LIF instrument with the same design as that used in this study was deployed in several field campaigns in China with the addition of a chemical titration device to separate ambient OH radicals from interferences, showing at maximum an interference of 10 % during daytime (Tan et al., 2017, 2018, 2019).
Results from the simulation chamber experiments are used to investigate the impact on the global distribution of the OH regeneration efficiency due to either radical reactions with NO or isomerization reactions by implementing the detailed isoprene chemistry as derived in this study within the M2 model (Table S7). The global atmospheric chemistry model EMAC (Jöckel et al., 2010) was applied, including a modified version of the Mainz Organic Mechanism (MOM) (Sander et al., 2019) that represents an advanced isoprene oxidation mechanism with a complexity comparable to the MCM.
In regions with low NO concentrations, OH regeneration by HO2 + NO is suppressed but compensated for by the OH regenerated from RO2 radical isomerization reactions. These reactions globally have the largest impact in the tropics due to high isoprene concentrations and high temperatures (Peeters et al., 2014). The inclusion of OH regeneration routes gives an OH regeneration efficiency that is at least 60 % globally over all land masses covered with vegetation (Fig. 8). As a consequence, in areas where isoprene is the most important reactant for the OH radicals, the concentration at the surface is enhanced by more than a factor of 3 compared to model predictions neglecting RO2 isomerization reactions (Fig. S3). Several studies showing the global impact of isomerization reactions were performed (Bates and Jacob, 2019; Møller et al., 2019; Müller et al., 2019), all showing, similarly to the results within this study, an enhanced concentration of the OH radical, in particular at the tropics where high concentrations of isoprene and high temperatures can be observed. A detailed comparison between the different models is not straightforward as they all contain different sets of reactions and are performed with different model parameters (e.g. NOx and isoprene emissions). Three published studies are based on the isoprene Caltech mechanism (Table 2), which underestimates the measured OH radical concentration from this study due to a low yield of the formation of the Z-δ-RO2 radical (Fig. 4). In addition, the model used in the study by Müller et al. (2019) includes large yields towards HPALD (0.75) as suggested from the study by Berndt et al. (2019), which is different from the other models. Thus, one additional global model test run within this work was performed with a yield of HPALD of 0.75 and of di-HPCARP-RO2 of 0.25 (comparable to M3) to verify if the alternative branching ratio would result in significant differences. However, no change in the expected OH radical concentration was observed, in agreement with the model runs for the chamber experiments. However, for small oxygenated volatile organic compounds (OVOCs) like formaldehyde, formic acid, methanol, glyoxal, methyl glyoxal, hydroxyacetone, and peroxy acetyl nitrate, a change of up to 30 % was found at the ground level in isoprene-dominated regions. In the same regions, the change in the CO concentration was less than 5 %.
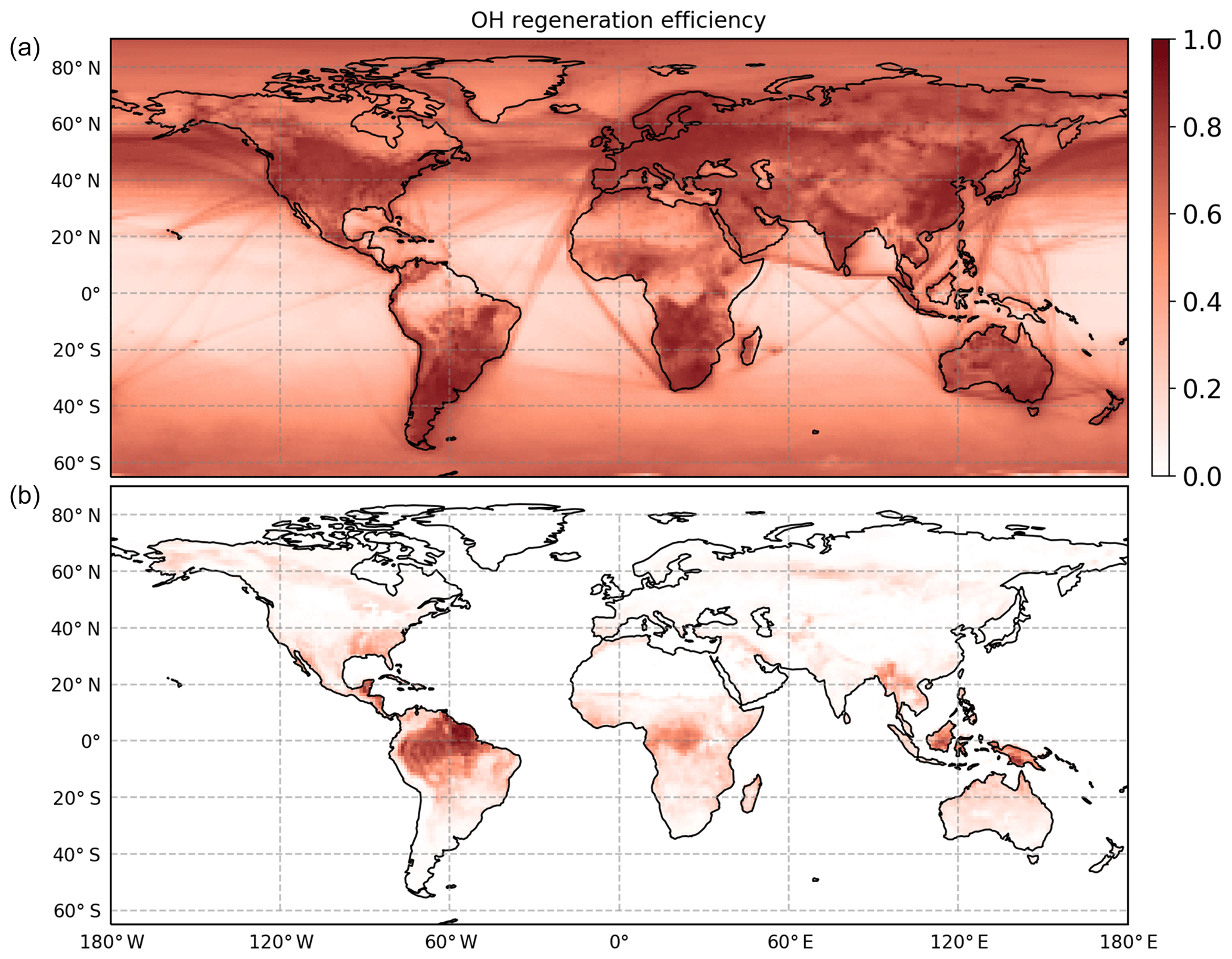
Figure 8Global model of the OH regeneration efficiency at the surface (M2). (a) The OH regeneration efficiency when considering only the reaction between HO2 radicals with NO and with O3. (b) Including the remaining contributions by isomerization reactions and the photolysis of HPALD. Isomerization reactions are very efficient in recycling the OH radicals and thus maintaining the oxidation capacity of the atmosphere in environments characterized by high isoprene and low NO.
7.1 Yield of di-HPCARP-RO2 versus HPALD
The LIM1 mechanism by Peeters et al. (2014) proposed a low ratio of HPALD to di-HPCARP-RO2 formation, in agreement with the Teng et al. (2017) experiments. Since then, the experimental study of Berndt et al. (2019) found a much higher HPALD yield, with small contributions only of di-HPCARP-RO2. The HPALD to di-HPCARP-RO2 ratio is governed by the chemistry of the HOO-hydroxy-allyl radicals formed after the dominant 1,6-H shift in the Z-δ-1,4-ISOPOO radicals (Fig. S4). The Müller et al. (2019) rationalization of the high HPALD yield by Berndt et al. (2019) is based mostly on the stereoselectivity of the Z-δ-1,4-ISOPOO H migration and the subsequent O2 addition, in connection with which new theoretical work (Müller et al., 2019) showed a comparatively high barrier for internal rotation of the HOC•H–C=C moiety in the HOO-hydroxy-allyl radicals due to the partial double bond. The nascent stereospecificity ( versus ; see Fig. S4 box A) then remains essentially unchanged throughout the subsequent chemistry of the HOO-hydroxy-allyl radicals; this is contrary to earlier assumptions in Peeters et al. (2014). HPALD can be formed rapidly from both stereoisomers by O2 addition on the HOC•H– radical carbon (Fig. S4 box D), followed by fast HO2 elimination. Di-HPCARP-RO2 is formed only from the -HOO-hydroxy-allyl radicals (Fig. S4 box E), as the fast enol H migration requires the geometric proximity of the –OH and –OO• groups.
Müller et al. (2019) suggest, based on the stereospecific chemistry, that the E′-enol-peroxy radicals formed from O2 addition on C4 of the -HOO-hydroxy-allyl radicals (Fig. S4 box B) will solely undergo re-dissociation, as well as the subsequent re-addition of O2 on either of the radical sites of the -HOO-hydroxy-allyl radical, until “indirect” (Müller et al., 2019) HPALD is formed. Thus, the high yield of HPALD is explained based on its formation from all -HOO-hydroxy-allyl radicals and part of the -HOO-hydroxy-allyl radicals, whereas di-HPCARP-RO2 is only formed from part of the radicals.
While this mechanism can indeed lead to numerical agreement with the Berndt et al. yields, the argumentation is not based on actual quantitative theoretical work on each reaction step and may thus be unable to discriminate between alternative mechanisms or yields in this subtle, complex chemistry; in this paragraph, we briefly examine a few aspects of the mechanism that warrant further investigation. Different yields would be obtained if the stereospecific yields (determined based on minimum energy pathways) are affected by non-statistical dynamics induced by chemical activation or post-barrier energy release. Likewise, the site specificity of O2 addition, based on radical spin densities as a first-order approximation rather than on characterizations of the addition TS, carries a large uncertainty. The rate of re-dissociation of the HOO-enol-peroxy adducts is suggested by Müller et al. (2019) to be very similar to the initial Z- and E-δ-OH-peroxy radicals from isoprene. However, H scrambling between the –OOH and –OO• groups (see Fig. S4 box C) is expected to be orders of magnitude faster (Miyoshi, 2011), leading to an equilibrated population in which the terminal peroxy radical has no access to a rapid re-dissociation channel, leading to lower overall phenomenological rate coefficients for re-dissociation. Without characterization of the impact of the terminal peroxy radical in the bulk rate coefficient it is difficult to assess whether sufficient re-dissociation and/or re-addition events can occur on the experimental timescale to ensure complete reconversion to HPALD. H scrambling also allows access to enol reaction pathways ignored by Müller et al. (2019). We examined ring closure and H-migration reactions for the HOCH=C(CH3)CH2CH2OO• proxy molecule, predicting a ring closure rate coefficient of s−1 (see Fig. S2) that dominates re-dissociation. The impact of the additional –OOH group in HOCH=C(CH3)CH(OOH)CH2OO• on the ring closure rate is hard to estimate without computational work, and these proxy results may thus not be applicable to the isoprene chemistry. At the very least, one expects significant differences between the case I and II rate coefficients. However, even a slower rate of ring closure would disrupt the reaction flow as laid out by Müller et al. (2019) and affect the predicted HPALD yield.
At this moment, there are contradictory experimental data on the HPALD vs. di-HPCARP-RO2 yield (Berndt, 2012; Teng et al., 2017; Berndt et al., 2019). The authors acknowledge that the reasons behind the disagreement are not clear. The earlier mechanism by Peeters et al. (2014) was used to rationalize the low HPALD observations of Teng et al. (2017) but did not appropriately account for stereospecificity. The Müller et al. (2019) mechanism is compatible with the high HPALD yield by Berndt et al. (2019), but it is based partially on mechanistic argumentations that may not be compatible with new and existing quantitative theoretical work despite the excellent apparent numerical agreement with experiments. We must stress that none of the aforementioned considerations listed here on the Müller et al. (2019) reaction scheme have the strength by themselves to invalidate the proposed mechanism and are mostly indications that caveats apply when implementing this scheme in chemical models. In this complex chemistry, with subtle competition between many channels, we feel there is as yet not enough quantitative theoretical work to claim theory-based support for either experimental yield.
It is therefore not possible from this study to unequivocally determine the correct yield of HPALD versus di-HPCARP-RO2, highlighting the need for further studies measuring their degradation products to pinpoint their yield of formation.
7.2 Fate of HPALD and di-hydroperoxy carbonyl compounds
HPALD is assumed to photolyse at a relatively fast rate ( s−1 at a 30∘ solar zenith angle), producing CO and OH radicals among other trace gases (Wolfe et al., 2012; Liu et al., 2017). In addition to photolysis it will react with OH radicals, for which MCMv3.3.1 includes three site-specific reactions with a total rate of cm3 s−1 (Wolfe et al., 2012). This reaction can compete with photolysis when OH radical concentrations reach 4×106 cm−3 ( s−1), but the product distribution has not yet been measured. The fate of the di-hydroperoxy carbonyl compounds (DHPMEK and DHPMPAL in the MCMv3.3.1 and MVK3OOH4OOH and MACR2OOH3OOH in the Caltech mechanism), formed from the aldehyde shift of the di-HPCARP-RO2, is more uncertain and within the M2 model they are predicted in relatively large concentrations (∼1010 cm−3). In the MCMv3.3.1 model, the two di-hydroperoxy carbonyl compounds degrade either by reacting with OH radicals, regenerating the OH radical, or by photolysis. Only two of the possible five site-specific reactions (Jenkin et al., 2018) are included, with a total rate coefficient of the order of cm3 s−1. Within the MCMv3.3.1 this is the main loss path for these species as photolysis is slower than s−1 and does not compete with the pseudo-first-order rate coefficient for the ambient concentration of OH radicals. A recent theoretical study by Liu et al. (2018) suggests, based on a calculation for the proxy molecule 2-hydroperoxypropanal, a much faster rate for the photolysis (∼1 to s−1 at a 30∘ solar zenith angle) with a yield of ∼20 % OH radicals. At an ambient concentration of OH radicals lower than 4×106 cm−3, photolysis would then become the dominant path. The Caltech mechanism, based on Liu et al. (2018), then has photolysis as the only degradation path for the di-hydroperoxy carbonyl compounds. As long as the yield of HPALD remains uncertain, it is difficult to assess the importance of these reactions. This underlines the need for additional studies on the degradation of di-hydroperoxy carbonyl compounds and HPALD as they can have, locally, a large impact on the type of oxygenated products obtained.
Photo-oxidation experiments on isoprene, the globally dominant biogenic volatile organic compound emitted, were performed in the atmospheric simulation chamber SAPHIR for a range of NO mixing ratios to explore the importance of the isomerization reaction for OH radical regeneration. Measurements of OH reactivity, OH, and radical concentrations, and other important trace gases were compared to results from different model calculations all based on a state-of-the-art chemical mechanistic model (MCMv3.3.1) (Jenkin et al., 2015).
It was found that the MCMv3.3.1 for isoprene degradation initiated by OH radicals is not able to reproduce the measured trace gas concentrations in the experiments despite the inclusion of the isomerization reaction for isoprene-RO2 following the LIM1 mechanism for NO mixing ratios <0.2 ppbv. Large discrepancies are observed, in particular for OH radicals, with a ratio of modelled to measured OH of 0.7±0.07 and of almost a factor of 2 for the sum of MVK, MACR, and ISOPOOHs (all isomers).
Summarizing the theoretical analysis, we find that the main fate of di-HPCARP-RO2-I is migration of the aldehyde H atom followed by rapid CO loss, leading to an unstable α-OOH alkyl radical that will eliminate an OH radical (Vereecken et al., 2004), forming DHP-MVK. The rate-limiting reaction is the aldehyde H migration, with an effective rate coefficient k(300 K) s−1. Alternative reaction channels are found to be uncompetitive. The mechanism leading to these results is significantly more complex than originally proposed (Peeters et al., 2014), with a rate coefficient significantly higher than the original 0.1 s−1 estimate and possibly a contribution of thermalized reactions of the tri-HPACYL intermediate. However, the nett product formation remains identical to that incorporated in the mechanisms by Peeters et al. (2014) and Wennberg et al. (2018) and implemented in e.g. the Master Chemical Mechanism v3.3.1 (Jenkin et al., 2015). Extrapolating these results to the di-HPCARP-RO2-II radicals by accounting for the expected barrier difference due to the different position of the methyl group, we find there is competition between CO elimination versus O2 addition in the tri-HPACYL-II radicals formed, especially when assuming the absence of chemical activation.
The kinetic aspects controlling the impact of the 1,6-H shift of Z-δ-RO2 on the regeneration of OH radicals and the production of oxygenated products were carefully checked based on what is available in the literature (Table 2). It was found that the best agreement between measured and modelled trace gases is observed when up to 50 % of the isoprene-RO2 conformers (weighted by their abundance) are isomerized. This is achieved when including within the MCMv3.3.1 a faster rate coefficient (3.6 and 0.4 s−1 at 298 K for OH addition on C4 and C1, respectively; Fig. 1) for the 1,6-H shift of the Z-δ-RO2 radical based on a recent experimental study (Teng et al., 2017). These changes result in a phenomenological bulk isomerization rate in agreement with what can be obtained from the LIM1 study by Peeters et al. (2014). Large uncertainties remain regarding the relative yield of HPALD and di-HPCARP-RO2 following the 1,6-H shift of Z-δ-RO2 radicals. Within this study, no meaningful differences between the results of different model calculations could be observed when the yield of HPALD was varied from 0.4 to 0.75. Both HPALD and di-HPCARP-RO2 produce OH radicals and CO in a relatively short timescale (less than 1 h). Therefore, as long as the 1,6-H shift is fast and there are sufficient Z-δ-RO2 radicals undergoing isomerization, the measurements from this study are not sensitive to the HPALD to di-HPCARP-RO2 ratio.
A detailed study of the path contribution to the OH radical regeneration highlights how, for NO mixing ratios <0.2 ppbv, ∼50 % of the OH radical is regenerated from the products following the 1,6-H shift: the photolysis of HPALD and aldehyde shift of the di-HPCARP-RO2. These processes help maintain the OH radical regeneration efficiency up to 0.5 in environments with low NO mixing ratios wherein regeneration via the reaction of HO2 with NO becomes less important. For environments in which the NO concentration is higher, regeneration via HO2 plus NO dominates (>75 %), and even models not including isomerization reactions are able to reproduce the measured trace gases. The observed OH radical regeneration efficiency in this chamber study at low NO mixing ratios study is, however, lower than what is observed and needed in the field to explain the measured OH radical concentrations in isoprene-dominated environments (Rohrer et al., 2014).
A semi-explicit global model which includes the chemistry highlighted in this study shows how isomerization helps maintain an OH regeneration efficiency up to 0.6 globally. In the Amazon at the ground level, the inclusion of isomerization reactions increases the OH radical concentrations up to a factor of 3, although this has no relevant impact on the global budget of methane or CO.
The data from the experiments in the SAPHIR chamber used in this work are available on the EUROCHAMP data home page (https://data.eurochamp.org/, last access: 1 October 2019, EUROCHAMP, 2019).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-3333-2020-supplement.
HF and AH designed the experiments. AN analysed the data, performed the box model simulations, and wrote the paper together with HF. LV did the theoretical calculations. SR and DT performed the global model simulations. All other co-authors participated in data collection and experiment operations, and all co-authors participated in paper discussion.
The authors declare that they have no conflict of interest.
This article is part of the special issue “Simulation chambers as tools in atmospheric research (AMT/ACP/GMD inter-journal SI)”. It is not associated with a conference.
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (SARLEP grant agreement no. 681529). The authors gratefully acknowledge the computing time granted through JARA-HPC on the supercomputer JURECA at Forschungszentrum Jülich Centre, 2018.
This research has been supported by the European
Research Council (SARLEP (grant no. 681529)), the European Commission, and
the H2020 Research Infrastructures (EUROCHAMP-2020 (grant no. 730997)).
The article processing charges for this open-access
publication were covered by a Research
Centre of the Helmholtz Association.
This paper was edited by Dwayne Heard and reviewed by three anonymous referees.
Alecu, I. M., Zheng, J., Zhao, Y., and Truhlar, D. G.: Computational Thermochemistry: Scale Factor Databases and Scale Factors for Vibrational Frequencies Obtained from Electronic Model Chemistries, J. Chem. Theory Comput., 6, 2872–2887, https://doi.org/10.1021/ct100326h, 2010.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006.
Bao, J. L., Zheng, J., Alecu, I. M., Lynch, B. J., Zhao, Y., and Truhlar, D. G.: DDatabase of Frequency Scale Factors for Electronic Model Chemistries (Version 3 Beta 2), available at: http://comp.chem.umn.edu/freqscale/index.html (last access: June 2018), 2017.
Bates, K. H. and Jacob, D. J.: A new model mechanism for atmospheric oxidation of isoprene: global effects on oxidants, nitrogen oxides, organic products, and secondary organic aerosol, Atmos. Chem. Phys., 19, 9613–9640, https://doi.org/10.5194/acp-19-9613-2019, 2019.
Baulch, D. L., Bowman, C. T., Cobos, C. J., Cox, R. A., Just, T., Kerr, J. A., Pilling, M. J., Stocker, D., Troe, J., Tsang, W., Walker, R. W., and Warnatz, J.: Evaluated Kinetic Data for Combustion Modeling: Supplement II, J. Phys. Chem. Ref. Data, 34, p. 757, https://doi.org/10.1063/1.1748524, 2005.
Berndt, T.: Formation of carbonyls and hydroperoxyenals (HPALDs) from the OH radical reaction of isoprene for low-NOx conditions: influence of temperature and water vapour content, J. Atmos. Chem., 69, 253–272, https://doi.org/10.1007/s10874-012-9245-2, 2012.
Berndt, T., Hyttinen, N., Herrmann, H., and Hansel, A.: First oxidation products from the reaction of hydroxyl radicals with isoprene for pristine environmental conditions, Comm. Chem., 2, p. 21, https://doi.org/10.1038/s42004-019-0120-9, 2019.
Blitz, M. A., Heard, D. E., and Pilling, M. J.: OH formation from CH3CO+O2: a convenient experimental marker for the acetyl radical, Chem. Phys. Lett., 365, 374–379, https://doi.org/10.1016/S0009-2614(02)01484-7, 2002.
Bohn, B. and Zilken, H.: Model-aided radiometric determination of photolysis frequencies in a sunlit atmosphere simulation chamber, Atmos. Chem. Phys., 5, 191–206, https://doi.org/10.5194/acp-5-191-2005, 2005.
Bohn, B., Rohrer, F., Brauers, T., and Wahner, A.: Actinometric measurements of NO2 photolysis frequencies in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 5, 493–503, https://doi.org/10.5194/acp-5-493-2005, 2005.
Carr, S. A., Glowacki, D. R., Liang, C.-H., Baeza-Romero, M. T., Blitz, M. A., Pilling, M. J., and Seakins, P. W.: Experimental and Modeling Studies of the Pressure and Temperature Dependences of the Kinetics and the OH Yields in the Acetyl + O2 Reaction, J. Phys. Chem. A, 115, 1069–1085, https://doi.org/10.1021/jp1099199, 2011.
Crounse, J. D., Teng, A., and Wennberg, P. O.: Experimental constrains on the distribution and fate of peroxy radicals formed in the reactions of isoprene + OH + O2 presented at the Atmospheric Chemical Mechanisms: Simple Models – Real world Complexities, Universtiy of California, Davis, USA, 10–12 December 2014.
Crounse, J. D., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Peroxy radical isomerization in the oxidation of isoprene, Phys. Chem. Chem. Phys., 13, 13607–13613, https://doi.org/10.1039/c1cp21330j, 2011.
Da Silva, G., Graham, C., and Wang, Z.-F.: Unimolecular β-hydroxyperoxy radical decomposition with OH recycling in the photochemical oxidation of isoprene, Environ. Sci. Technol., 44, 250–256, https://doi.org/10.1021/es900924d, 2010.
Dorn, H.-P., Brandenburger, U., Brauers, T., and Hausmann, M.: A New In Situ Laser Long-Path Absorption Instrument for the Measurement of Tropospheric OH Radicals, J. Atmos. Sci., 52, 3373–3380, https://doi.org/10.1175/1520-0469(1995)052<3373:anisll>2.0.co;2, 1995.
Dunning, T. H.: Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 90, 1007–1023, https://doi.org/10.1063/1.456153, 1989.
EUROCHAMP: EUROCHAMP Data Centre, https://data.eurochamp.org/, last access: 1 October 2019.
Fuchs, H., Bohn, B., Hofzumahaus, A., Holland, F., Lu, K. D., Nehr, S., Rohrer, F., and Wahner, A.: Detection of HO2 by laser-induced fluorescence: calibration and interferences from RO2 radicals, Atmos. Meas. Tech., 4, 1209–1225, https://doi.org/10.5194/amt-4-1209-2011, 2011.
Fuchs, H., Dorn, H.-P., Bachner, M., Bohn, B., Brauers, T., Gomm, S., Hofzumahaus, A., Holland, F., Nehr, S., Rohrer, F., Tillmann, R., and Wahner, A.: Comparison of OH concentration measurements by DOAS and LIF during SAPHIR chamber experiments at high OH reactivity and low NO concentration, Atmos. Meas. Tech., 5, 1611–1626, https://doi.org/10.5194/amt-5-1611-2012, 2012.
Fuchs, H., Hofzumahaus, A., Rohrer, F., Bohn, B., Brauers, T., Dorn, H. P., Haseler, R., Holland, F., Kaminski, M., Li, X., Lu, K., Nehr, S., Tillmann, R., Wegener, R., and Wahner, A.: Experimental evidence for efficient hydroxyl radical regeneration in isoprene oxidation, Nat. Geosci., 6, 1023–1026, https://doi.org/10.1038/Ngeo1964, 2013.
Fuchs, H., Acir, I.-H., Bohn, B., Brauers, T., Dorn, H.-P., Häseler, R., Hofzumahaus, A., Holland, F., Kaminski, M., Li, X., Lu, K., Lutz, A., Nehr, S., Rohrer, F., Tillmann, R., Wegener, R., and Wahner, A.: OH regeneration from methacrolein oxidation investigated in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 14, 7895–7908, https://doi.org/10.5194/acp-14-7895-2014, 2014.
Fuchs, H., Tan, Z., Hofzumahaus, A., Broch, S., Dorn, H.-P., Holland, F., Künstler, C., Gomm, S., Rohrer, F., Schrade, S., Tillmann, R., and Wahner, A.: Investigation of potential interferences in the detection of atmospheric ROx radicals by laser-induced fluorescence under dark conditions, Atmos. Meas. Tech., 9, 1431–1447, https://doi.org/10.5194/amt-9-1431-2016, 2016.
Fuchs, H., Novelli, A., Rolletter, M., Hofzumahaus, A., Pfannerstill, E. Y., Kessel, S., Edtbauer, A., Williams, J., Michoud, V., Dusanter, S., Locoge, N., Zannoni, N., Gros, V., Truong, F., Sarda-Esteve, R., Cryer, D. R., Brumby, C. A., Whalley, L. K., Stone, D., Seakins, P. W., Heard, D. E., Schoemaecker, C., Blocquet, M., Coudert, S., Batut, S., Fittschen, C., Thames, A. B., Brune, W. H., Ernest, C., Harder, H., Muller, J. B. A., Elste, T., Kubistin, D., Andres, S., Bohn, B., Hohaus, T., Holland, F., Li, X., Rohrer, F., Kiendler-Scharr, A., Tillmann, R., Wegener, R., Yu, Z., Zou, Q., and Wahner, A.: Comparison of OH reactivity measurements in the atmospheric simulation chamber SAPHIR, Atmos. Meas. Tech., 10, 4023–4053, https://doi.org/10.5194/amt-10-4023-2017, 2017.
Guenther, A., Karl, T., Harley, P., Wiedinmyer, C., Palmer, P. I., and Geron, C.: Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature), Atmos. Chem. Phys., 6, 3181–3210, https://doi.org/10.5194/acp-6-3181-2006, 2006.
Hofzumahaus, A., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C.-C., Fuchs, H., Holland, F., Kita, K., Kondo, Y., Li, X., Lou, S., Shao, M., Zeng, L., Wahner, A., and Zhang, Y.: Amplified trace gas removal in the troposphere, Science, 324, 1702–1704, https://doi.org/10.1126/science.1164566, 2009.
Holland, F., Hofzumahaus, A., Schafer, R., Kraus, A., and Patz, H. W.: Measurements of OH and HO2 radical concentrations and photolysis frequencies during BERLIOZ, J. Geophys. Res.-Atmos., 108, 8246, https://doi.org/10.1029/2001jd001393, 2003.
Hornbrook, R. S., Crawford, J. H., Edwards, G. D., Goyea, O., Mauldin III, R. L., Olson, J. S., and Cantrell, C. A.: Measurements of tropospheric HO2 and RO2 by oxygen dilution modulation and chemical ionization mass spectrometry, Atmos. Meas. Tech., 4, 735–756, https://doi.org/10.5194/amt-4-735-2011, 2011.
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015.
Jenkin, M. E., Valorso, R., Aumont, B., Rickard, A. R., and Wallington, T. J.: Estimation of rate coefficients and branching ratios for gas-phase reactions of OH with aliphatic organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 18, 9297–9328, https://doi.org/10.5194/acp-18-9297-2018, 2018.
Jenkin, M. E., Khan, M. A. H., Shallcross, D. E., Bergström, R., Simpson, D., Murphy, K. L. C., and Rickard, A. R.: The CRI v2.2 reduced degradation scheme for isoprene, Atmos. Environ., 212, 172–182, https://doi.org/10.1016/j.atmosenv.2019.05.055, 2019.
Jöckel, P., Kerkweg, A., Pozzer, A., Sander, R., Tost, H., Riede, H., Baumgaertner, A., Gromov, S., and Kern, B.: Development cycle 2 of the Modular Earth Submodel System (MESSy2), Geosci. Model Dev., 3, 717–752, https://doi.org/10.5194/gmd-3-717-2010, 2010.
Jordan, A., Haidacher, S., Hanel, G., Hartungen, E., Mark, L., Seehauser, H., Schottkowsky, R., Sulzer, P., and Mark, T. D.: A high resolution and high sensitivity proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS), Int. J. Mass Spectrom., 286, 122–128, https://doi.org/10.1016/j.ijms.2009.07.005, 2009.
Jorgensen, S., Knap, H. C., Otkjaer, R. V., Jensen, A. M., Kjeldsen, M. L. H., Wennberg, P. O., and Kjaergaard, H. G.: Rapid Hydrogen Shift Scrambling in Hydroperoxy-Substituted Organic Peroxy Radicals, J. Phys. Chem. A, 120, 266–275, https://doi.org/10.1021/acs.jpca.5b067613, 2016.
Kaminski, M., Fuchs, H., Acir, I.-H., Bohn, B., Brauers, T., Dorn, H.-P., Häseler, R., Hofzumahaus, A., Li, X., Lutz, A., Nehr, S., Rohrer, F., Tillmann, R., Vereecken, L., Wegener, R., and Wahner, A.: Investigation of the β-pinene photooxidation by OH in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 17, 6631–6650, https://doi.org/10.5194/acp-17-6631-2017, 2017.
Karl, M., Dorn, H.-P., Holland, F., Koppmann, R., Poppe, D., Rupp, L., Schaub, A., and Wahner, A.: Product study of the reaction of OH radicals with isoprene in the atmosphere simulation chamber SAPHIR, J. Atmos. Chem., 55, 167–187, https://doi.org/10.1007/s10874-006-9034-x, 2006.
Knap, H. C. and Jorgensen, S.: Rapid Hydrogen Shift Reactions in Acyl Peroxy Radicals, J. Phys. Chem. A, 121, 1470–1479, https://doi.org/10.1021/acs.jpca.6b12787, 2017.
Kubistin, D., Harder, H., Martinez, M., Rudolf, M., Sander, R., Bozem, H., Eerdekens, G., Fischer, H., Gurk, C., Klüpfel, T., Königstedt, R., Parchatka, U., Schiller, C. L., Stickler, A., Taraborrelli, D., Williams, J., and Lelieveld, J.: Hydroxyl radicals in the tropical troposphere over the Suriname rainforest: comparison of measurements with the box model MECCA, Atmos. Chem. Phys., 10, 9705–9728, https://doi.org/10.5194/acp-10-9705-2010, 2010.
Lelieveld, J., Butler, T. M., Crowley, J. N., Dillon, T. J., Fischer, H., Ganzeveld, L., Harder, H., Lawrence, M. G., Martinez, M., Taraborrelli, D., and Williams, J.: Atmospheric oxidation capacity sustained by a tropical forest, Nature, 452, 737–740, https://doi.org/10.1038/nature06870, 2008.
Levy, H.: Photochemistry of the Troposphere, in: Advances in Photochemistry, John Wiley & Sons, Inc., 369–524, 1974.
Lew, M. M., Dusanter, S., and Stevens, P. S.: Measurement of interferences associated with the detection of the hydroperoxy radical in the atmosphere using laser-induced fluorescence, Atmos. Meas. Tech., 11, 95–109, https://doi.org/10.5194/amt-11-95-2018, 2018.
Lindinger, W., Hansel, A., and Jordan, A.: On-line monitoring of volatile organic compounds at pptv levels by means of proton-transfer-reaction mass spectrometry (PTR-MS) – Medical applications, food control and environmental research, Int. J. Mass. Spectrom., 173, 191–241, https://doi.org/10.1016/S0168-1176(97)00281-4, 1998.
Liu, Z., Nguyen, V. S., Harvey, J., Müller, J.-F., and Peeters, J.: Theoretically derived mechanisms of HPALD photolysis in isoprene oxidation, Phys. Chem. Chem. Phys., 19, 9096–9106, https://doi.org/10.1039/C7CP00288B, 2017.
Liu, Z., Nguyen, V. S., Harvey, J., Muller, J.-F., and Peeters, J.: The photolysis of [small alpha]-hydroperoxycarbonyls, Phys. Chem. Chem. Phys., 20, 6970–6979, https://doi.org/10.1039/C7CP08421H, 2018.
Lou, S., Holland, F., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C. C., Fuchs, H., Häseler, R., Kita, K., Kondo, Y., Li, X., Shao, M., Zeng, L., Wahner, A., Zhang, Y., Wang, W., and Hofzumahaus, A.: Atmospheric OH reactivities in the Pearl River Delta – China in summer 2006: measurement and model results, Atmos. Chem. Phys., 10, 11243–11260, https://doi.org/10.5194/acp-10-11243-2010, 2010.
Mao, J., Ren, X., Zhang, L., Van Duin, D. M., Cohen, R. C., Park, J.-H., Goldstein, A. H., Paulot, F., Beaver, M. R., Crounse, J. D., Wennberg, P. O., DiGangi, J. P., Henry, S. B., Keutsch, F. N., Park, C., Schade, G. W., Wolfe, G. M., Thornton, J. A., and Brune, W. H.: Insights into hydroxyl measurements and atmospheric oxidation in a California forest, Atmos. Chem. Phys., 12, 8009–8020, https://doi.org/10.5194/acp-12-8009-2012, 2012.
Méreau, R., Rayez, M.-T., Rayez, J.-C., Caralp, F., and Lesclaux, R.: Theoretical study on the atmospheric fate of carbonyl radicals: kinetics of decomposition reactions, Phys. Chem. Chem. Phys., 3, 4712–4717, https://doi.org/10.1039/B105824J, 2001.
Miyoshi, A.: Systematic computational study on the unimolecular reactions of alkylperoxy (RO2), hydroperoxyalkyl (QOOH), and hydroperoxyalkylperoxy (O2QOOH) radicals, J. Phys. Chem. A, 115, 3301–3325, https://doi.org/10.1021/jp112152n, 2011.
Miyoshi, A.: Molecular size dependent falloff rate constants for the recombination reactions of alkyl radicals with O2 and implications for simplified kinetics of alkylperoxy radicals, Int. J. Chem. Kinet., 44, 59–74, https://doi.org/10.1002/kin.20623, 2012.
Møller, K. H., Bates, K. H., and Kjaergaard, H. G.: The importance of peroxy radical hydrogen-shift reactions in atmospheric isoprene oxidation, J. Phys. Chem. A, 123, 920–932, https://doi.org/10.1021/acs.jpca.8b10432, 2019.
Müller, J.-F., Stavrakou, T., and Peeters, J.: Chemistry and deposition in the Model of Atmospheric composition at Global and Regional scales using Inversion Techniques for Trace gas Emissions (MAGRITTE v1.1) – Part 1: Chemical mechanism, Geosci. Model Dev., 12, 2307–2356, https://doi.org/10.5194/gmd-12-2307-2019, 2019.
Novelli, A., Hens, K., Tatum Ernest, C., Kubistin, D., Regelin, E., Elste, T., Plass-Dülmer, C., Martinez, M., Lelieveld, J., and Harder, H.: Characterisation of an inlet pre-injector laser-induced fluorescence instrument for the measurement of atmospheric hydroxyl radicals, Atmos. Meas. Tech., 7, 3413–3430, https://doi.org/10.5194/amt-7-3413-2014, 2014.
Novelli, A., Bohn, B., Dorn, H.-P., Hofzumahaus, A., Holland, F., Li, X., Kaminski, M., Yu, Z., Rosanka, S., Reimer, D., Gkatzelis, G. I., Taraborrelli, D., Vereecken, L., Rohrer, F., Tillmann, R., Wegener, R., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: The atmosphere of a tropical forest simulated in a chamber: experiments, theory and global significance of OH regeneration in isoprene oxidation, iCACGP-IGAC 2018 Conference, 25–29 September 2018, Takamatsu, Japan, 2018a.
Novelli, A., Kaminski, M., Rolletter, M., Acir, I.-H., Bohn, B., Dorn, H.-P., Li, X., Lutz, A., Nehr, S., Rohrer, F., Tillmann, R., Wegener, R., Holland, F., Hofzumahaus, A., Kiendler-Scharr, A., Wahner, A., and Fuchs, H.: Evaluation of OH and HO2 concentrations and their budgets during photooxidation of 2-methyl-3-butene-2-ol (MBO) in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 18, 11409–11422, https://doi.org/10.5194/acp-18-11409-2018, 2018b.
Park, J., Jongsma, C. G., Zhang, R., and North, S. W.: OH/OD Initiated Oxidation of Isoprene in the Presence of O2 and NO, J. Phys. Chem. A, 108, 10688–10697, https://doi.org/10.1021/jp040421t, 2004.
Peeters, J.: Interactive comment on “The MCM v3.3. degradation scheme for isoprene” by M. E. Jenkin et al., Atmos. Chem. Phys. Discuss., 15, C2486–C2486, 2015.
Peeters, J. and Müller, J.-F.: HOx radical regeneration in isoprene oxidation via peroxy radical isomerisations. II: experimental evidence and global impact, Phys. Chem. Chem. Phys., 12, 14227–14235, https://doi.org/10.1039/c0cp00811g, 2010.
Peeters, J., Nguyen, T. L., and Vereecken, L.: HOx radical regeneration in the oxidation of isoprene, Phys. Chem. Chem. Phys., 11, 5935–5939, https://doi.org/10.1039/b908511d, 2009.
Peeters, J., Müller, J.-F., Stavrakou, T., and Nguyen, V. S.: Hydroxyl radical recycling in isoprene oxidation driven by hydrogen bonding and hydrogen tunneling: the upgraded LIM1 mechanism, J. Phys. Chem. A, 118, 8625–8643 https://doi.org/10.1021/jp5033146, 2014.
Poppe, D., Brauers, T., Dorn, H.-P., Karl, M., Mentel, T., Schlosser, E., Tillmann, R., Wegener, R., and Wahner, A.: OH-initiated degradation of several hydrocarbons in the atmosphere simulation chamber SAPHIR, J. Atmos. Chem., 57, 203–214, https://doi.org/10.1007/s10874-007-9065-y, 2007.
Praske, E., Otkjær, R. V., Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric autoxidation is increasingly important in urban and suburban North America, P. Natl. Acad. Sci. USA, 115, 64–69, https://doi.org/10.1073/pnas.1715540115, 2018.
Praske, E., Otkjær, R. V., Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., and Wennberg, P. O.: Intramolecular Hydrogen Shift Chemistry of Hydroperoxy-Substituted Peroxy Radicals, J. Phys. Chem. A, 123, 590–600, https://doi.org/10.1021/acs.jpca.8b09745, 2019.
Purvis, G. D. and Bartlett, R. J.: A full coupled-cluster singles and doubles model: The inclusion of disconnected triples, J. Chem. Phys., 76, 1910, https://doi.org/10.1063/1.443164, 1982.
Ren, X., Olson, J. R., Crawford, J. H., Brune, W. H., Mao, J., Long, R. B., Chen, Z., Chen, G., Avery, M. A., Sachse, G. W., Barrick, J. D., Diskin, G. S., Huey, L. G., Fried, A., Cohen, R. C., Heikes, B., Wennberg, P. O., Singh, H. B., Blake, D. R., and Shetter, R. E.: HOx chemistry during INTEX-A 2004: Observation, model calculation, and comparison with previous studies, J. Geophys. Res.-Atmos., 113, D05310, https://doi.org/10.1029/2007jd009166, 2008.
Rickly, P. and Stevens, P. S.: Measurements of a potential interference with laser-induced fluorescence measurements of ambient OH from the ozonolysis of biogenic alkenes, Atmos. Meas. Tech., 11, 1–16, https://doi.org/10.5194/amt-11-1-2018, 2018.
Ridley, B. A., Grahek, F. E., and Walega, J. G.: A small, high-sensitivity, medium-response ozone detector suitable for measurements from light aircraft, J. Atmos. Ocean. Tech., 9, 142–148, https://doi.org/10.1175/1520-0426(1992)009, 1992.
Rivera-Rios, J. C., Nguyen, T. B., Crounse, J. D., Jud, W., St. Clair, J. M., Mikoviny, T., Gilman, J. B., Lerner, B. M., Kaiser, J. B., de Gouw, J., Wisthaler, A., Hansel, A., Wennberg, P. O., Seinfeld, J. H., and Keutsch, F. N.: Conversion of hydroperoxides to carbonyls in field and laboratory instrumentation: observational bias in diagnosing pristine versus anthropogenically-controlled atmospheric chemistry, Geophys. Res. Lett., 41, 8645–8651, https://doi.org/10.1002/2014gl061919, 2014.
Roeckner, E., Brokopf, R., Esch, M., Giorgetta, M., Hagemann, S., Kornblueh, L., Manzini, E., Schlese, U., and Schulzweida, U.: Sensitivity of Simulated Climate to Horizontal and Vertical Resolution in the ECHAM5 Atmosphere Model, J. Climate, 19, 3771–3791, https://doi.org/10.1175/jcli3824.1, 2006.
Rohrer, F., Bohn, B., Brauers, T., Brüning, D., Johnen, F.-J., Wahner, A., and Kleffmann, J.: Characterisation of the photolytic HONO-source in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 5, 2189–2201, https://doi.org/10.5194/acp-5-2189-2005, 2005.
Rohrer, F., Lu, K., Hofzumahaus, A., Bohn, B., Brauers, T., Chang, C.-C., Fuchs, H., Häseler, R., Holland, F., Hu, M., Kita, K., Kondo, Y., Li, X., Lou, S., Oebel, A., Shao, M., Zeng, L., Zhu, T., Zhang, Y., and Wahner, A.: Maximum efficiency in the hydroxyl-radical-based self-cleansing of the troposphere, Nat. Geosci., 7, 559, https://doi.org/10.1038/ngeo2199, 2014.
Sander, R., Baumgaertner, A., Gromov, S., Harder, H., Jöckel, P., Kerkweg, A., Kubistin, D., Regelin, E., Riede, H., Sandu, A., Taraborrelli, D., Tost, H., and Xie, Z.-Q.: The atmospheric chemistry box model CAABA/MECCA-3.0, Geosci. Model Dev., 4, 373–380, https://doi.org/10.5194/gmd-4-373-2011, 2011.
Sander, R., Baumgaertner, A., Cabrera-Perez, D., Frank, F., Gromov, S., Grooß, J.-U., Harder, H., Huijnen, V., Jöckel, P., Karydis, V. A., Niemeyer, K. E., Pozzer, A., Riede, H., Schultz, M. G., Taraborrelli, D., and Tauer, S.: The community atmospheric chemistry box model CAABA/MECCA-4.0, Geosci. Model Dev., 12, 1365–1385, https://doi.org/10.5194/gmd-12-1365-2019, 2019.
Schlosser, E., Bohn, B., Brauers, T., Dorn, H.-P., Fuchs, H., Häseler, R., Hofzumahaus, A., Holland, F., Rohrer, F., Rupp, L., Siese, M., Tillmann, R., and Wahner, A.: Intercomparison of two hydroxyl radical measurement techniques at the atmosphere simulation chamber SAPHIR, J. Atmos. Chem., 56, 187–205, https://doi.org/10.1007/s10874-006-9049-3, 2007.
Schlosser, E., Brauers, T., Dorn, H.-P., Fuchs, H., Häseler, R., Hofzumahaus, A., Holland, F., Wahner, A., Kanaya, Y., Kajii, Y., Miyamoto, K., Nishida, S., Watanabe, K., Yoshino, A., Kubistin, D., Martinez, M., Rudolf, M., Harder, H., Berresheim, H., Elste, T., Plass-Dülmer, C., Stange, G., and Schurath, U.: Technical Note: Formal blind intercomparison of OH measurements: results from the international campaign HOxComp, Atmos. Chem. Phys., 9, 7923–7948, https://doi.org/10.5194/acp-9-7923-2009, 2009.
Sehested, J., Christensen, L. K., Nielsen, O. J., and Wallington, T. J.: Absolute rate constants for F + CH3CHO and CH3CHO + O2, relative rate study of CH3CHO + NO, and the product distribution of the F + CH3CHO reaction, Int. J. Chem. Kinet., 30, 913–921, https://doi.org/10.1002/(SICI)1097-4601(1998)30:12<913::AID-KIN6>3.0.CO;2-5, 1998.
Sindelarova, K., Granier, C., Bouarar, I., Guenther, A., Tilmes, S., Stavrakou, T., Müller, J.-F., Kuhn, U., Stefani, P., and Knorr, W.: Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years, Atmos. Chem. Phys., 14, 9317–9341, https://doi.org/10.5194/acp-14-9317-2014, 2014.
St. Clair, J. M., Rivera-Rios, J. C., Crounse, J. D., Knap, H. C., Bates, K. H., Teng, A. P., Jørgensen, S., Kjaergaard, H. G., Keutsch, F. N., and Wennberg, P. O.: Kinetics and Products of the Reaction of the First-Generation Isoprene Hydroxy Hydroperoxide (ISOPOOH) with OH, The J. Phys. Chem. A, 120, 1441–1451, https://doi.org/10.1021/acs.jpca.5b06532, 2016.
Tan, D., Faloona, I., Simpas, J. B., Brune, W., Shepson, P. B., Couch, T. L., Sumner, A. L., Carroll, M. A., Thornberry, T., Apel, E., Riemer, D., and Stockwell, W.: HOx budgets in a deciduous forest: Results from the PROPHET summer 1998 campaign, J. Geophys. Res., 106, 24407–24427, https://doi.org/10.1029/2001jd900016, 2001.
Tan, Z., Fuchs, H., Lu, K., Hofzumahaus, A., Bohn, B., Broch, S., Dong, H., Gomm, S., Häseler, R., He, L., Holland, F., Li, X., Liu, Y., Lu, S., Rohrer, F., Shao, M., Wang, B., Wang, M., Wu, Y., Zeng, L., Zhang, Y., Wahner, A., and Zhang, Y.: Radical chemistry at a rural site (Wangdu) in the North China Plain: observation and model calculations of OH, HO2 and RO2 radicals, Atmos. Chem. Phys., 17, 663–690, https://doi.org/10.5194/acp-17-663-2017, 2017.
Tan, Z., Rohrer, F., Lu, K., Ma, X., Bohn, B., Broch, S., Dong, H., Fuchs, H., Gkatzelis, G. I., Hofzumahaus, A., Holland, F., Li, X., Liu, Y., Liu, Y., Novelli, A., Shao, M., Wang, H., Wu, Y., Zeng, L., Hu, M., Kiendler-Scharr, A., Wahner, A., and Zhang, Y.: Wintertime photochemistry in Beijing: observations of ROx radical concentrations in the North China Plain during the BEST-ONE campaign, Atmos. Chem. Phys., 18, 12391–12411, https://doi.org/10.5194/acp-18-12391-2018, 2018.
Tan, Z., Lu, K., Hofzumahaus, A., Fuchs, H., Bohn, B., Holland, F., Liu, Y., Rohrer, F., Shao, M., Sun, K., Wu, Y., Zeng, L., Zhang, Y., Zou, Q., Kiendler-Scharr, A., Wahner, A., and Zhang, Y.: Experimental budgets of OH, HO2, and RO2 radicals and implications for ozone formation in the Pearl River Delta in China 2014, Atmos. Chem. Phys., 19, 7129–7150, https://doi.org/10.5194/acp-19-7129-2019, 2019.
Taraborrelli, D., Lawrence, M. G., Crowley, J. N., Dillon, T. J., Gromov, S., Groß, C. B. M., Vereecken, L., and Lelieveld, J.: Hydroxyl radical buffered by isoprene oxidation over tropical forests, Nat. Geosci., 5, 190–193, 2012.
Teng, A. P., Crounse, J. D., and Wennberg, P. O.: Isoprene peroxy radical dynamics, J. Am. Chem. Soc., 139, 5367–5377, https://doi.org/10.1021/jacs.6b12838, 2017.
Vereecken, L. and Peeters, J.: The 1,5-H-shift in 1-butoxy: A case study in the rigorous implementation of transition state theory for a multirotamer system, J. Chem. Phys., 119, 5159–5170, https://doi.org/10.1063/1.1597479, 2003.
Vereecken, L., Nguyen, T. L., Hermans, I., and Peeters, J.: Computational study of the stability of α-hydroperoxyl- or α-alkylperoxyl substituted alkyl radicals, Chem. Phys. Lett., 393, 432–436, https://doi.org/10.1016/j.cplett.2004.06.076, 2004.
Wang, S., Riva, M., Yan, C., Ehn, M., and Wang, L.: Primary formation of highly oxidized multifunctional products in the OH-Initiated oxidation of Isoprene: a combined theoretical and experimental study, Environ. Sci. Technol., 52, 12255–12264, https://doi.org/10.1021/acs.est.8b02783, 2018.
Wegener, R., Brauers, T., Koppmann, R., Rodríguez Bares, S., Rohrer, F., Tillmann, R., Wahner, A., Hansel, A., and Wisthaler, A.: Simulation chamber investigation of the reactions of ozone with short-chained alkenes, J. Geophys. Res.-Atmos., 112, D13301, https://doi.org/10.1029/2006JD007531, 2007.
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-phase reactions of isoprene and its major oxidation products, Chem. Rev., 118, 3337–3390, https://doi.org/10.1021/acs.chemrev.7b00439, 2018.
Whalley, L. K., Edwards, P. M., Furneaux, K. L., Goddard, A., Ingham, T., Evans, M. J., Stone, D., Hopkins, J. R., Jones, C. E., Karunaharan, A., Lee, J. D., Lewis, A. C., Monks, P. S., Moller, S. J., and Heard, D. E.: Quantifying the magnitude of a missing hydroxyl radical source in a tropical rainforest, Atmos. Chem. Phys., 11, 7223–7233, https://doi.org/10.5194/acp-11-7223-2011, 2011.
Whalley, L. K., Blitz, M. A., Desservettaz, M., Seakins, P. W., and Heard, D. E.: Reporting the sensitivity of laser-induced fluorescence instruments used for HO2 detection to an interference from RO2 radicals and introducing a novel approach that enables HO2 and certain RO2 types to be selectively measured, Atmos. Meas. Tech., 6, 3425–3440, https://doi.org/10.5194/amt-6-3425-2013, 2013.
Wolfe, G. M., Crounse, J. D., Parrish, J. D., St. Clair, J. M., Beaver, M. R., Paulot, F., Yoon, T. P., Wennberg, P. O., and Keutsch, F. N.: Photolysis, OH reactivity and ozone reactivity of a proxy for isoprene-derived hydroperoxyenals (HPALDs), Phys. Chem. Chem. Phys., 14, 7276–7286, https://doi.org/10.1039/C2CP40388A, 2012.
Xing, L., Lucas, J., Wang, Z., Wang, X., and Truhlar, D. G.: Hydrogen shift isomerizations in the kinetics of the second oxidation mechanism of alkane combustion, Reactions of the hydroperoxypentylperoxy OOQOOH radical, Combust. Flame, 197, 88–101, https://doi.org/10.1016/j.combustflame.2018.07.013, 2018.
Zhao, Y. and Truhlar, D. G.: The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 120, 215–241, https://doi.org/10.1007/s00214-007-0310-x, 2008.
- Abstract
- Introduction
- Methods
- Theoretical work on isoprene di-HPCARP-RO2-I
- Comparison of measured trace gases with model calculations
- Modelled contributions to the measured OH radical regeneration efficiency
- Global impact
- Remaining uncertainties
- Summary and conclusion
- Code and data availability
- Author contributions
- Competing interests
- Special issue statement
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Methods
- Theoretical work on isoprene di-HPCARP-RO2-I
- Comparison of measured trace gases with model calculations
- Modelled contributions to the measured OH radical regeneration efficiency
- Global impact
- Remaining uncertainties
- Summary and conclusion
- Code and data availability
- Author contributions
- Competing interests
- Special issue statement
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement