the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 07 Jun 2019
| 07 Jun 2019
Emission of volatile halogenated organic compounds over various Dead Sea landscapes
Moshe Shechner
Alex Guenther
Robert Rhew
Asher Wishkerman
Qian Li
Donald Blake
Gil Lerner
Volatile halogenated organic compounds (VHOCs), such as methyl halides (CH3X; X is Br, Cl and I) and very short-lived halogenated substances (VSLSs; bromoform – CHBr3, dibromomethane – CH2Br2, bromodichloromethane – CHBrCl2, trichloroethylene – C2HCl3, chloroform – CHCl3 – and dibromochloromethane – CHBr2Cl) are well known for their significant influence on ozone concentrations and oxidation capacity of the troposphere and stratosphere and for their key role in aerosol formation. Insufficient characterization of the sources and the emission rate of VHOCs limits our ability to understand and assess their impact in both the troposphere and stratosphere. Over the last two decades, several natural terrestrial sources for VHOCs, including soil and vegetation, have been identified, but our knowledge of emission rates from these sources and their responses to changes in ambient conditions remains limited. Here we report measurements of the mixing ratios and fluxes of several chlorinated and brominated VHOCs from different landscapes and natural and agricultural vegetated sites at the Dead Sea during different seasons. Fluxes were generally positive (emission into the atmosphere), corresponding to elevated mixing ratios, but were highly variable. Fluxes (and mixing ratios) for the investigated VHOCs ranged as follows: CHBr3 from −79 to 187 nmol m−2 d−1 (1.9 to 22.6 pptv), CH2Br2 from −55 to 71 nmol m−2 d−1 (0.7 to 19 pptv), CHBr2Cl from −408 to 768 nmol m−2 d−1 (0.4 to 11 pptv), CHBrCl2 from −29 to 45 nmol m−2 d−1 (0.5 to 9.6 pptv), CHCl3 from −577 to 883 nmol m−2 d−1 (15 to 57 pptv), C2HCl3 from −74 to 884 nmol m−2 d−1 (0.4 to 11 pptv), methyl chloride (CH3Cl) from -5300 to 10,800 nmol m−2 d−1 (530 to 730 pptv), methyl bromide (CH3Br) from −111 to 118 nmol m−2 d−1 (7.5 to 14 pptv) and methyl iodide (CH3I) from −25 to 17 nmol m−2 d−1 (0.4 to 2.8 pptv). Taking into account statistical uncertainties, the coastal sites (particularly those where soil is mixed with salt deposits) were identified as sources of all VHOCs, but this was not statistically significant for CHCl3. Further away from the coastal area, the bare soil sites were sources for CHBrCl2, CHBr2Cl, CHCl3, and probably also for CH2Br2 and CH3I, and the agricultural sites were sources for CHBr3, CHBr2Cl and CHBrCl2. In contrast to previous reports, we also observed emissions of brominated trihalomethanes, with net molar fluxes ordered as follows: CHBr2Cl > CHCl3 > CHBr3 > CHBrCl2 and lowest positive flux incidence for CHCl3 among all trihalomethanes; this finding can be explained by the soil's enrichment with Br. Correlation analysis, in agreement with recent studies, indicated common controls for the emission of CHBr2Cl and CHBrCl2 and likely also for CHBr3. There were no indications for correlation of the brominated trihalomethanes with CHCl3. Also in line with previous reports, we observed elevated emissions of CHCl3 and C2HCl3 from mixtures of soil and different salt-deposited structures; the flux correlations between these compounds and methyl halides (particularly CH3I) suggested that at least CH3I is also emitted via similar mechanisms or is subjected to similar controls. Overall, our results indicate elevated emission of VHOCs from bare soil under semiarid conditions. Along with other recent studies, our findings point to the strong emission potential of a suite of VHOCs from saline soils and salt lakes and call for additional studies of emission rates and mechanisms of VHOCs from saline soils and salt lakes.
- Article
(4267 KB) -
Supplement
(1522 KB) - BibTeX
- EndNote





Volatile halogenated organic compounds (VHOCs), such as methyl halides (CH3X; X is Br, Cl and I) and very short-lived halogenated substances (VSLSs; lifetime < 6 months), contribute substantially to the loading of tropospheric and lower stratospheric reactive halogen species containing Cl, Br or I, and their oxides (Carpenter et al., 2013, 2014; Derendorp et al., 2012). Reactive halogen species, in turn, lead to ozone (O3) destruction, changes in atmospheric oxidation capacity and radiative forcing (Simpson et al., 2015). Depletion of O3 in the stratosphere is associated with damage to biological tissues owing to an increase in transmittance of UVB radiation (Rousseaux et al., 1999). In the troposphere, O3 destruction is of great importance, given that O3 is toxic to humans, plants and animals; is a greenhouse gas; and plays a key role in the oxidation capacity of the atmosphere.
The lifetimes of VHOCs vary significantly (see summary in Table S1 in the Supplement), which in turn affects their influence in both the troposphere and the stratosphere. Owing to their relatively short lifetimes (< 6 months), the transport of VSLSs to the stratosphere occurs primarily in the tropics, where deep convection is frequent. Brominated VSLSs originate primarily from the ocean, whereas chlorinated VSLSs, except for chloroform (CHCl3) and chloroethane, originate primarily from anthropogenic sources (Carpenter et al., 2014). Methyl iodide (CH3I), having a relatively short lifetime, is also classified as a VSLS and contributes significantly to tropospheric O3 destruction in the marine boundary layer (MBL; Carpenter et al., 2014) and also, indirectly, to the formation of cloud condensation nuclei (O'Dowd et al., 2002). It is now well established that emission of brominated (e.g., bromoform – CHBr3, methylene bromide – CH2Br2 – and dibromochloromethane – CHBr2Cl) and iodinated (e.g., CH3I) VSLSs tends to be much greater in coastal areas than in the open ocean (Carpenter et al., 2000, 2009; Liu et al., 2011; Bondu et al., 2008; Manley and Dastoor, 1988; Quack and Wallace, 2004), since in the former, they can also be emitted from macroalgae under oxidative stress at low tide (Pedersen et al., 1996). The ocean is also a major source of methyl bromide (CH3Br) and a significant (∼19 %) source of methyl chloride (CH3Cl; Carpenter et al., 2014), as they originate from phytoplankton, bacteria and detritus.
Despite the numerous efforts made in recent years to evaluate halocarbon budgets, uncertainties still exist concerning the strengths of both their sources and their sinks. The budgets of CH3Br and CH3Cl are unbalanced, with sinks outweighing sources by ∼32 % and ∼17 %, respectively (Carpenter et al., 2014). Uncertainties in the global budgets of naturally occurring VSLSs are large, with discrepancies having a factor of ∼2–3 between top-down and bottom-up emission inventories (Carpenter et al., 2014). This results largely from poor characterization of emission sources (Warwick et al., 2006; Hossaini et al., 2013; Ziska et al., 2013).
Studies over the past few decades have clearly demonstrated that terrestrial sources also constitute a major fraction of the atmospheric budget for both methyl halides and VSLSs (Carpenter et al., 2014). Many terrestrial plants have been identified as sources of CH3Cl (Yokouchi et al., 2007), and the results of recent modeling indicate that about 55 % of the global sources of CH3Cl originate from tropical lands (Xiao et al., 2010; Carpenter et al., 2014). It has also been suggested that natural terrestrial sources of CH3Br, especially emissions from terrestrial vegetation, must account for a large part of the missing sources (Gebhardt et al., 2008; Yassaa et al., 2009; Warwick et al., 2006; Gan et al., 1998; Yokouchi et al., 2002; Moore, 2006; Rhew et al., 2001; Wishkerman et al., 2008), and emissions have been observed from peatlands, wetlands, salt marshes, shrublands, forests and some cultivated crops (Gan et al., 1998; Varner et al., 1999; Lee-Taylor and Holland, 2000). CHCl3 has also been found to be emitting from various terrestrial sources, including rice, soil, tundra, forest floor and different types of microorganisms, such as fungi and termites (see Dimmer et al., 2001 and Rhew et al., 2008).
The importance of VHOC emission from soil, sediments and salt lake deposits has been recently recognized (see Kotte et al., 2012; Ruecker et al., 2014, and references therein). For example, Keppler et al. (2000) revealed natural abiotic emission of CH3Br, CH3Cl and CH3I as well as additional chlorinated VHOCs from soil and sediments harboring an oxidant such as Fe(III), halides, or organic matter (OM), while Weissflog et al. (2005) found that salt lake sediments can be a source for several C1 and C2 chlorinated species, including CHCl3 and trichloroethylene (C2HCl3), induced by halobacteria in the presence of dissolved Fe. Huber et al. (2009) identified abiotic natural emission of trihalomethanes from soil, including CHCl3, bromodichloromethane (CHBrCl2) and CHBr2Cl, induced by oxidation of OM by Fe(III) and hydrogen peroxide, while Hoekstra et al. (1998) identified natural emission of CHBr3 following enrichment of the soil by potassium bromide. In addition, Carpenter et al. (2005) identified CHBr3 emission from peatland or another terrestrial source at Mace Head (in Ireland). Albers et al. (2017) revealed that CHCl3, CHBrCl2 and potentially also other trihalomethanes can be emitted from soils, probably induced by hydrolysis of trihaloacetyl compounds. Several other studies have reported strong emissions of CH3Cl, CH3Br and CH3I from coastal marsh vegetation and to a lesser extent from the marsh soil (Rhew et al., 2000, 2002, 2014; Wishkerman et al., 2008), with significant importance on a global scale (Deventer et al., 2018; Manley et al., 2006). In addition, peatland has been indicated as an important source for CH3Br, CH3Cl, CH3I and CHCl3 (Simmonds et al., 2010; Khan et al., 2012; Dimmer et al., 2001; Carpenter et al., 2005), and Sive et al. (2007) identified a globally significant source of CH3I from midlatitude vegetation and soil.
Accordingly, the need for improved understanding of VHOC emissions from saline environments and their potential importance on the global scale have been highlighted by recent studies (Weissflog et al., 2005; Kotte et al., 2012; Ruecker et al., 2014; Deventer et al., 2018). Moreover, due to global warming, saline environments are likely to become more prevalent (IPCC 2007; Ruecker et al., 2014; Jiao et al., 2018). The present study is aimed at improving our knowledge of the emission of VHOCs from salt lake environments by quantifying the flux and mixing ratios of methyl halides and halogenated VSLSs from different sites in the area of the Dead Sea.
The Dead Sea is unique because it is the lowest point on the Earth's surface, about 430 m below sea level, with water salinity 12 times higher and a ratio of bromide (Br−) to chloride (Cl−; ) that is 7.5 times higher than in normal ocean waters. Fast evaporation from the sea leads to a variety of newly exposed sea deposits. Despite the high salinity, emission of VHOCs via biotic processes at the Dead Sea is also potentially feasible. The unicellular green alga Dunaliella parva has been found to be active in Dead Sea water (Oren and Shilo, 1985), while additional bacteria and fungi that have been isolated from the sea could also potentially be active under the Dead Sea's extreme conditions (Oren et al., 2008; Jacob et al., 2017; Buchalo et al., 1998). Mycobiota, including fungi and biota, have also been detected in the Dead Sea's hypersaline soil and coastal sand (Pen-Mouratov et al., 2010; Kis-Papo et al., 2001; Jacob et al., 2017).
Studying the emission of VHOCs at the Dead Sea is also fundamental for understanding local surface O3-depletion events (Hebestreit et al., 1999; Tas et al., 2003, 2006; Matveev et al., 2001; Zingler and Platt, 2005) as well as mercury-depletion events (Tas et al., 2012; Obrist et al., 2011) in the boundary layer of this area. Emissions of brominated and iodinated VHOCs can potentially lead to formation of the reactive iodine and bromine species that are responsible for these processes.
Table 1Summary of VHOC samplings at the Dead Sea. Shown are the date, time and site name (and abbreviation) for the sample, sampling height, total number of samplings for each experiment and whether the sample could potentially have been influenced by emission from the seawater and by precipitation prior to sampling.
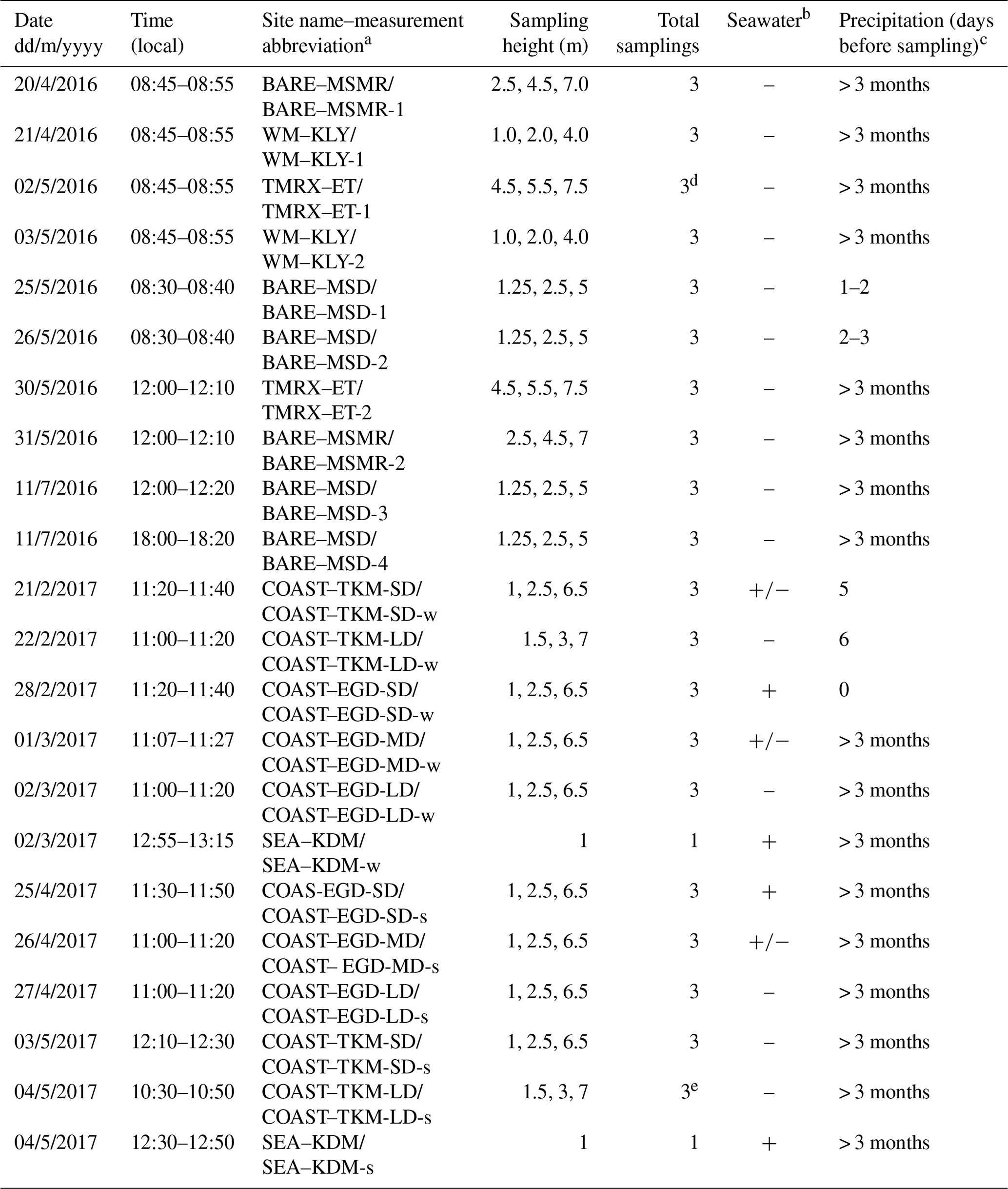
a The suffixes “s” and “w” refer to samples taken during the spring and winter, respectively. SD, MD and LD refer to relatively short, medium and long distance from the coastline, respectively (see Sect. 2.1). b “+”, “–” and “+/–” indicate that the samplings were, could not be or may be influenced by emission from the seawater, respectively. c Values indicate the number of days before sampling on which precipitation occurred. Additional abbreviations: MSD – Masada, MSMR – Mishmar, KLY – Kalya, ET – Ein Tamar, KDM – Kedem, EGD – Ein Gedi, BARE – bare soil site, COAST – coastal soil–salt mixture site, WM – agricultural cultivated watermelon site, TMRX – natural site with Tamarix – and SEA – sampling near the seawater (see Sect. 2.1.1). d Samples exclude one CH3Cl measurement in TMRX–ET-1 (see Sect. 2.1.2). e Samples exclude one measurement for all VHOCs (see Sect. 2.1.2).
2.1 Field measurements and sampling
Field measurements were taken at selected sites along the Dead Sea to measure the mixing ratios and evaluate the vertical flux of VHOCs over different land-use types, seasons and distance from the seawater, as summarized in Table 1. Soil samples from the various sites were analyzed, and meteorological measurements were performed in situ.
2.1.1 Measurement sites
All measurements were taken in the Dead Sea area. The Dead Sea's geographical position is between 31∘00′ N and 31∘50′ N at 35∘30′ E, about 430 m below sea level. It is located in a semiarid area, with mean daily maximum temperatures for summer and winter of ∼40 and ∼21 ∘C, respectively. The Dead Sea has low rates of freshwater inflow and precipitation (20–50 mm yr−1; Shafir and Alpert, 2010), while seawater evaporation rates are high, estimated at about 400 cm yr−1 (Alpert et al., 1997). As a result, the water salinity is 12 times higher than the average salinity of ocean water. Dead Sea water contains on average 5.6 g L−1 Br− and 225 g L−1 Cl− ( ≈0.025; Niemi et al., 1997), whereas normal ocean water contains 0.065 g L−1 Br− and 19 g L−1 Cl− ( ≈0.0034; Sverdrup et al., 1942). The main anthropogenic emission source in the area, apart from local transportation and a few small settlements, is the Dead Sea Works, a potash plant located to the south of most of the measurement sites (see Fig. 1). Agricultural fields, which are mostly concentrated in the north near Kalya, in the south near Ein Tamar and near Ein Gedi (see Fig. 1), are also potential sources for the emission of VHOCs in the area. To the best of our knowledge, there are no wastewater facilities near the Dead Sea area, which could otherwise also contribute to the emission of VHOCs such as CHCl3 and CHBr3.
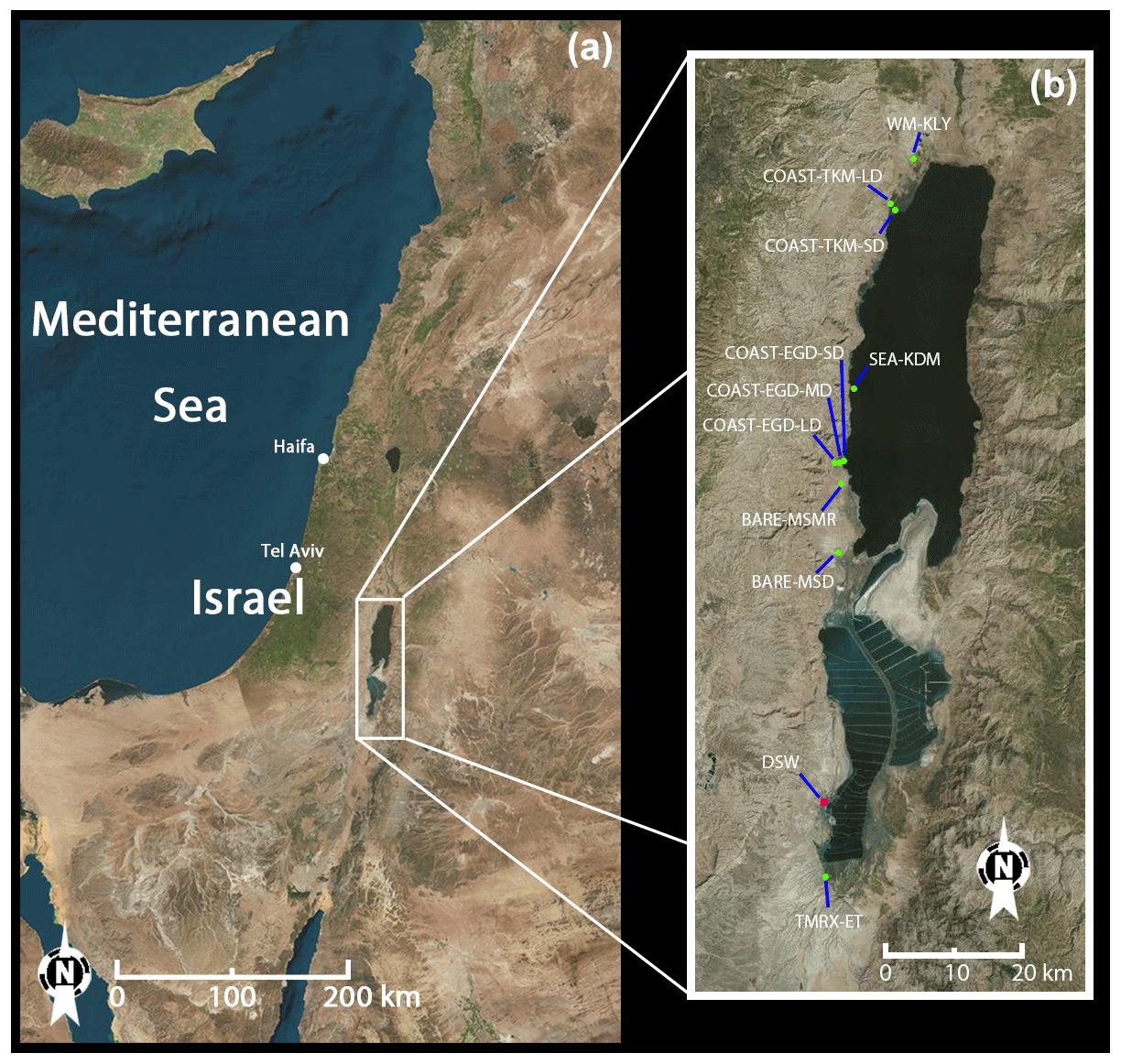
Figure 1Location and satellite image of the Dead Sea measurement sites (see Sect. 2.1.2) and Dead Sea Works (DSW). (a) Location of the Dead Sea. (b) Zoomed view of the area of the measurement sites.
All measurement sites were nearly flat, homogeneous and located either along or near the Dead Sea coast (see Fig. 1). Sites were classified according to surface cover: bare soil sites at Mishmar (BARE–MSMR) and at Masada (BARE–MSD), coastal sites that are mixtures of soil and salt deposits at Ein Gedi (COAST–EGD) and Tzukim (COAST–TKM), natural Tamarix vegetation at Ein Tamar (TMRX–ET), irrigated agricultural watermelon field at Kalya (WM–KLY), and seawater at Kedem (SEA–KDM). Note that at SEA–KDM, we did not evaluate fluxes. Based on in situ wind-direction measurements, the sampled air masses at SEA–KDM were transported over the seawater from the east (see Fig. 1) at least 1 h prior to sampling and during the sampling. To study the effect of distance from the seawater on emission rates, measurements at both COAST–EGD and COAST–TKM were taken at three and two different distances from the sea, respectively. The shorter, middle and longer distances from the seawater were termed, respectively, SD, MD and LD. Emission rates at both COAST–EGD and COAST–TKM could potentially be affected by distance from the seashore; there are several reasons for this, including changes across the sites in soil salt and water content and changes in density of the extremely sparse vegetation cover. In addition, depending on the local wind direction at COAST–TKM-SD and COAST–EGD-SD, direct emission and uptake from the seawater can potentially affect the samples.
In the following, we briefly describe the different measurement sites; additional information about the sites and measurements is provided in Table 1. BARE–MSMR has bare soil consisting of loess and a small fraction of drifted soil covered with small stones and extremely sparse vegetation and is located in a valley 1.5 km to the west of the Dead Sea shore. BARE–MSD has bare Hamada soil, with small stones and loess, and is located 2.1 km to the west of the Dead Sea. COAST–EGD-SD has dried-out bare saline soil, mixed with salty beds and rocks, with a small contribution of freshwater inflow. COAST–EGD-MD has a dried-out seabed of bare saline soil, mixed with salty beds and rocks, and is located 0.3 km west of the Dead Sea shore. COAST–EGD-LD is a dried-out seabed of loess saline bare soil, mixed with drifted soil, and is located 0.8 km from the Dead Sea shore. COAST–TKM-SD is wetted bare soil with salt deposits, groundwater inflow from the Dead Sea and minor (< 5 %) freshwater inflow lines covered with perennial grasses found in wetlands (e.g., Phragmites sp.) and is located about 0.5 km from the shore. COAST–TKM-LD is a flat rocky loess area about 1.5 km from the shore, with patchy salts and sparse mixed shallow vegetation, mostly small Atriplex sp., Tamarix sp. and Retama raetam. TMRX–ET is a moderately dense Tamarix grove, of 4–5 m average height, with an area of ∼2.25 km2, a 60 %–70 % vegetation cover fraction and sandy soil, located 1.7 km south of the southern tip of the Dead Sea evaporation ponds (see Fig. 1). Lastly, WM–KLY is a well-irrigated and flat 700 m×350 m agricultural field with cultivated watermelon surrounded by a larger agricultural area of ∼3 km2, located 2.5 km northwest of the Dead Sea shore (Fig. 1). The watermelon crop had an average height of ∼0.67 m and 95 %–99 % vegetation cover.
2.1.2 Field measurements and sampled air analysis
Air was sampled at each site by placing three different canisters at specified heights (see Table 1) along a meteorological tower. The samples were used to quantify the mixing ratios of different VHOCs in the air, and their corresponding fluxes were calculated by applying the flux-gradient method (see Stull, 1988; Maier and Schack-Kirchner, 2014; Meredith et al., 2014). By default, the differences in height between the canisters increased exponentially with height, considering the typical decrease in the vertical gradient of emitted species in the surface layer (Stull, 1988). All canisters were placed high enough above the ground to ensure that all sampling was performed within the inertial sublayer, except for the lowest canister at TMRX–ET. In all cases, the sample footprint fell inside the target fetch, except for the sampling at COAST–EGD, for which the sample footprint included a narrow strip of the seawater (estimated at about 40 % of the footprint). To minimize non-synchronized air sampling by the three canisters, we constructed a special sampling system that allows almost simultaneous filling of the canisters. For each sample, air was drawn into a 1.9 L stainless-steel canister via passive grab samplers (Restek Corporation, PA, USA), resulting in a sampling duration of 20 min and internal canister pressures higher than 600 Torr. Meteorological parameters, including temperature and relative humidity, wind speed and direction, and global solar radiation, were all continuously measured, starting at least 30 min before air sampling was initiated (summarized in Table S6). All canisters were sent to the Blake–Rowland group, University of California, Irvine, where they were analyzed by techniques similar to those described in Colman et al. (2001). Analyses were performed using gas chromatography combined with mass spectrometry, flame-ionization detection and electron-capture detection to quantify the air mixing ratios of CHBr3, C2HCl3, CH2Br2, CHBr2Cl, CHBrCl2, CHCl3, CH3I, CH3Br and CH3Cl. For all gases, accuracy ranged from 1 % to 10 %, and analytical precision ranged from 1 % to 5 % (see Table S2). Note that the lower-height canister analysis for COAST–TKM-LD-s and the mid-height canister analysis of TMRX–ET-1 indicated an outlier mixing ratio for all VHOCs and for CH3Cl, respectively (p ≪ 0.01; Grubbs test; Grubbs and Beck, 1972). We therefore excluded the lower-height COAST–TKM-LD-s measurement from all of our calculations and used only the lowest and highest canisters in the flux calculation for TMRX–ET-1, as indicated in all relevant figures and tables.
2.2 Vertical flux evaluation
The vertical flux, Fc, of a species, c, was evaluated according to the gradient approach using the vertical gradient of c, , and a constant, Kc:
Kc represents the rate of turbulent exchange in Eq. (1) and was evaluated on the basis of the Monin–Obukhov similarity theory (MOST) described by Lenschow (1995):
where u∗ is the friction velocity, K is the Von Kármán constant, Z is the measurement height and φc is a universal function of the dimensionless parameter ζ. According to MOST, vertical fluxes in the surface layer can be evaluated on the basis of the dimensionless length parameter, ζ, according to
where z, d and L are the vertical coordinate, zero displacement and the Monin–Obukhov length, respectively (Schmugge and André, 1991).
We relied on the commonly used assumption that φC is similar to φh for chemical species with a relatively long lifetime (Dearellano et al., 1995) and calculated φh using the following equation for the relationship between φh and ζ, which was found to be valid for (Dyer and Bradley, 1982; Yang et al., 2001):
We derived L from the Pasquill and Gifford stability class (Pasquill and Smith, 1971) and roughness length (z0) according to Golder (1972). z0 was evaluated based on the specific surface characteristics at each site using information provided by Jarraud (2008). The stability class was evaluated using the solar radiation and wind speed measured in situ (Gifford, 1976; Pasquill and Smith, 1971). u∗ was derived from the logarithmic wind profile according to MOST, using the following equation:
where u(z) is the wind speed at height z, and ψm is a correction for diabatic effect on momentum transport. Using the measured u at a height of 10 m, we calculated the wind speed at each measurement height according to Gualtieri and Secci (2011):
where ψm is calculated using
and
2.3 Soil analyses
Soil samples at each site were collected up to a depth of 5 cm during the summer, at least 3 months after any rain event in the Dead Sea area, to ensure no impact on the samples by recent drift and percolation. The samples were analyzed for Br, Cl, I, OM, moisture and Fe in the soil as well as for soil pH. Prior to halide quantification, extractions for each sample were prepared using HNO3 (BSI, 1990). Total Br and I were quantified using inductively coupled plasma mass spectrometry (ICP–MS). Total Cl was quantified by potentiometric titration against AgNO3.
To quantify Fe in the soil, microwave-assisted digestion with reverse aqua regia was used, and Fe concentration was determined by inductively coupled plasma optical emission spectrometry (ICP–OES). A batch of each sample (∼300 mg of dry soil) was digested in reverse aqua regia (HNO3 – 65 % – to HCl – 30 %; 3:1 mixture, v∕v). Digestion was allowed to proceed in quartz vessels using a Discover sample digestion system at high temperature and pressure (CEM Corporation, NC, USA). The vessels were cooled and the volume was brought to 20 mL with deionized water. Element concentrations were measured in clear solutions using high-resolution dual-view ICP–OES PlasmaQuant PQ 9000 Elite (Analytik Jena, Germany). The reported values represent the lower limit because the samples were not completely dissolved. Soil water content and OM were determined by weight loss under dry combustion at 105 and 400 ∘C, respectively. Soil pH was measured in 1:1 (v∕v) soil-to-water extracts with a model 420 pH meter (Thermo Orion, MA, USA).
3.1 VHOC flux and mixing ratio
Overall, the measurements at the Dead Sea boundary layer revealed higher mixing ratios for all investigated VHOCs than their expected levels at the Mediterranean Sea and Red Sea MBL, indicating higher local emissions from the Dead Sea area. No association was observed between the measured mixing ratios and the air masses flowing from the direction of the Dead Sea Works (see Sect. S5 for anthropogenic impact), a potash plant located to the northwest of the TMRX–ET site and to the south of all other measurement sites (see Fig. 1), that is the main anthropogenic source in the area under investigation. Furthermore, the correlation analysis (Table S5) revealed that only C2HCl3 was associated with C2Cl4, a well-known anthropogenic VHOC. The absence of any other associations suggested dominance of natural sources for the VHOCs in the studied area. The measured mixing ratios for the different species at the measurement sites are summarized and compared with mixing ratios from the MBL in Table S3 and in Fig. 2. The figure indicates that median mixing ratios measured at the Dead Sea were generally higher than the corresponding mixing ratios in the MBL. Our calculations suggest that the mixing ratios at the Dead Sea are higher by factors of 1.2–8.0 for brominated and chlorinated VSLSs and ∼1.5, 1.3 and 1.1 for CH3I, CH3Br and CH3Cl, respectively. It should be noted, however, that while Fig. 2 implies elevated VHOC emission from the Dead Sea, comparison of mean or median mixing ratios of VHOCs for the Dead Sea with those for the MBL is not straightforward, considering that VHOC mixing ratios in the MBL are sensitive to several factors, including season and latitude. Moreover, the measurement height can play a significant role in affecting the mixing ratios due to decreasing mixing ratios with height over areas where local emissions occur. Hence, we also compared the measured fluxes and mixing ratios with their corresponding values measured in coastal areas, where the highest mixing ratios in the MBL were generally measured due to stronger emissions. The measured mixing ratios and fluxes at the Dead Sea were in most cases comparable to or higher than in coastal areas.
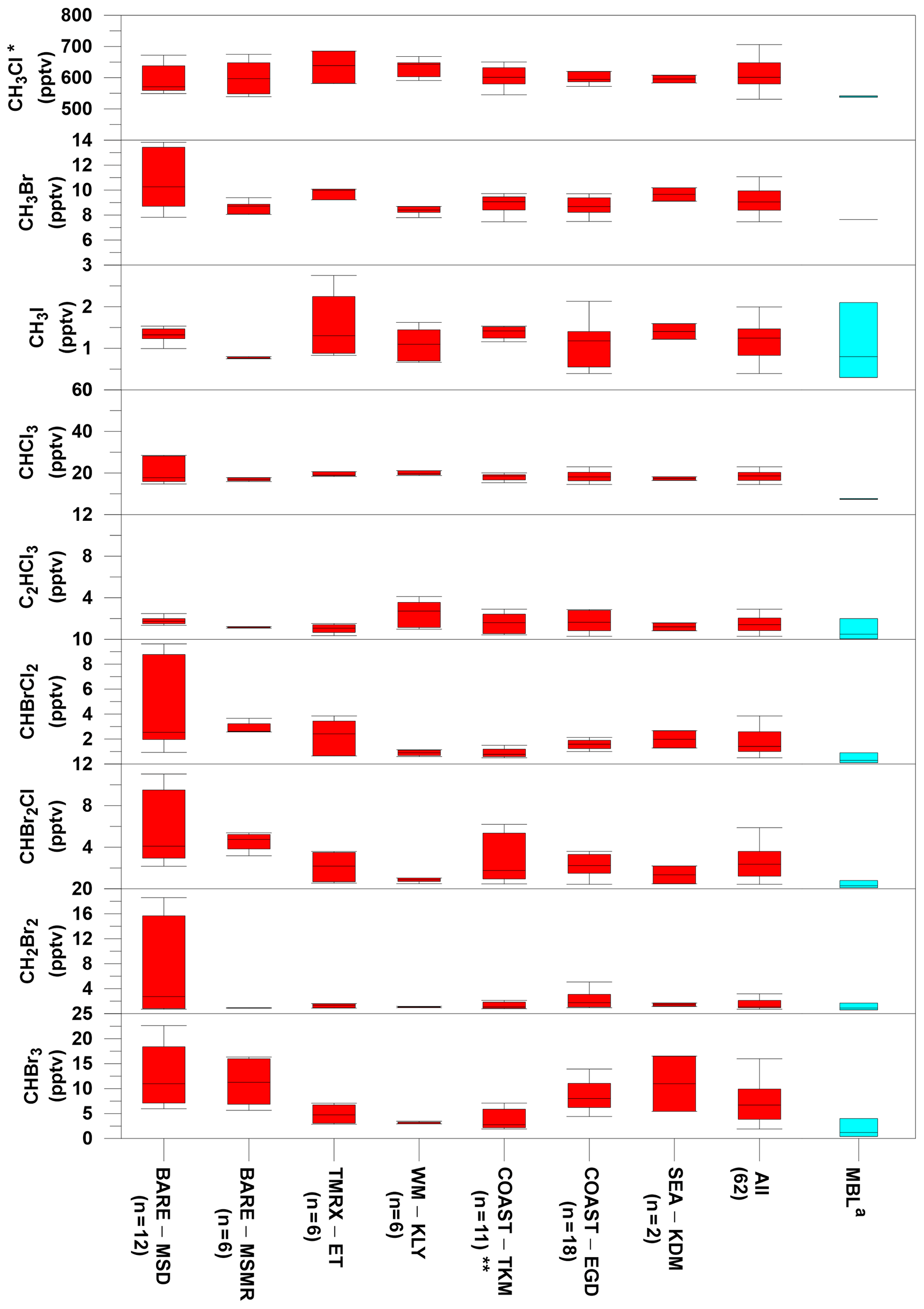
Figure 2Comparison of VHOC mixing ratios (in pptv) measured at the Dead Sea with their corresponding values at the marine boundary layer (MBL). For the Dead Sea sites, boxes indicate median, upper and lower quartiles, and bars show minimum and maximum VHOC mixing ratios (see Table 1 for site abbreviations; n specifies the number of samples for each site). For the MBL, boxes indicate the median, minimum and maximum mixing ratios reported by Carpenter et al. (2014). a Values for CH3Cl and CH3Br represent mean and range for 2012 based on flask measurements by the US National Oceanic and Atmospheric Administration (NOAA; http://www.esrl.noaa.gov/gmd/dv/site/, last access: 2019) and in situ measurements by the Advanced Global Atmospheric Gases Experiment (AGAGE; http://agage.eas.gatech.edu/, last access: 2019), which were performed at ground stations, not in all cases representing the MBL. * Related CH3Cl measurement excludes one sample at TMRX–ET-1 (see Sect. 2.1.2). Related measurements exclude one sample for all VHOCs (see Sect. 2.1.2).
Owing to their large contribution to stratospheric Br, CHBr3 and CH2Br2 are the most extensively studied VSLSs in the MBL (Hossaini et al., 2010). The mixing ratios of CHBr3 and CH2Br2 that we measured at the Dead Sea ranged from 1.9 to 22.6 pptv and from 0.7 to 18.6 pptv, respectively, higher than most of their reported mixing ratios in coastal areas where the highest mixing ratios have typically been measured. For example, Carpenter et al. (2009) reported elevated mixing ratios for CHBr3 and CH2Br2 along the eastern Atlantic coast, ranging from 1.9 to 4.9 and from 0.9 to 1.4 ppt, respectively, and Mohd Nadzir et al. (2014) reported mixing ratios of 0.82–5.25 pptv and 0.90–1.92 ppt for CHBr3 and CH2Br2, respectively, for several tropical coastal areas, including the Strait of Malacca, the South China Sea and the Sulu–Sulawesi Sea. Somewhat higher mixing ratios for CHBr3 have been measured in only a few locations, including some in coastal areas near New Hampshire (Zhou et al., 2008), San Cristóbal Island (Yokouchi et al., 2005; O'Brien et al., 2009), Cabo Verde (O'Brien et al., 2009), Borneo (Pyle et al., 2011), Cape Point (Kuyper et al., 2018; Butler et al., 2007) and at the Atmospheric Observing Station at Thompson Farm (TF) in New Hampshire, USA, during the summer (Zhou et al., 2005), whereas the range (and average) concentrations at those locations were 0.2–37.9 pptv (5.6–6.3), 4.2–43.6 pptv (14.2), 2.0–43.7 pptv (4.3–13.5), 0.2–60 pptv (1.3–1.7), 4.4–64.6 pptv (24.8) and 0.6–37.9 pptv (2.6–5.9), respectively. For CH2Br2, the corresponding mixing ratios were reported as 1.3–2.3, 0.5–4.1, 0.7–8.8 and 0.4–4.2 pptv in New Hampshire, San Cristóbal Island, Cabo Verde and TF, respectively, which are comparable with the mixing ratios measured at the Dead Sea.
Table 2VHOC fluxes for the different measurement sites. Shown is the measured flux (nmol m−2 d−1) obtained for the different measurements. Values in bold and in parentheses indicate that the related measurement site is a significant (p < 0.05) or non-significant (p > 0.15) net source or sink for the specific VHOC based on one-sample t test. Additional categories are defined below. These calculations assume COAST–EGD-SD and COAST–TKM-SD to be the same source (see Sect. 2.1.2). Also shown are the average flux (mean) and average positive flux (mean positive) for all species, as well as the percentage of incidence of positive flux (X) out of total measured fluxes, individually for each site and each VHOC (See Table 1 for abbreviations of the different measurement sites). All presented values, including mean, mean positive and X include only fluxes associated with p < 0.05 (bolded; S) and values associated with p≥0.05 (presented in parentheses; NS), based on one-sample t test.
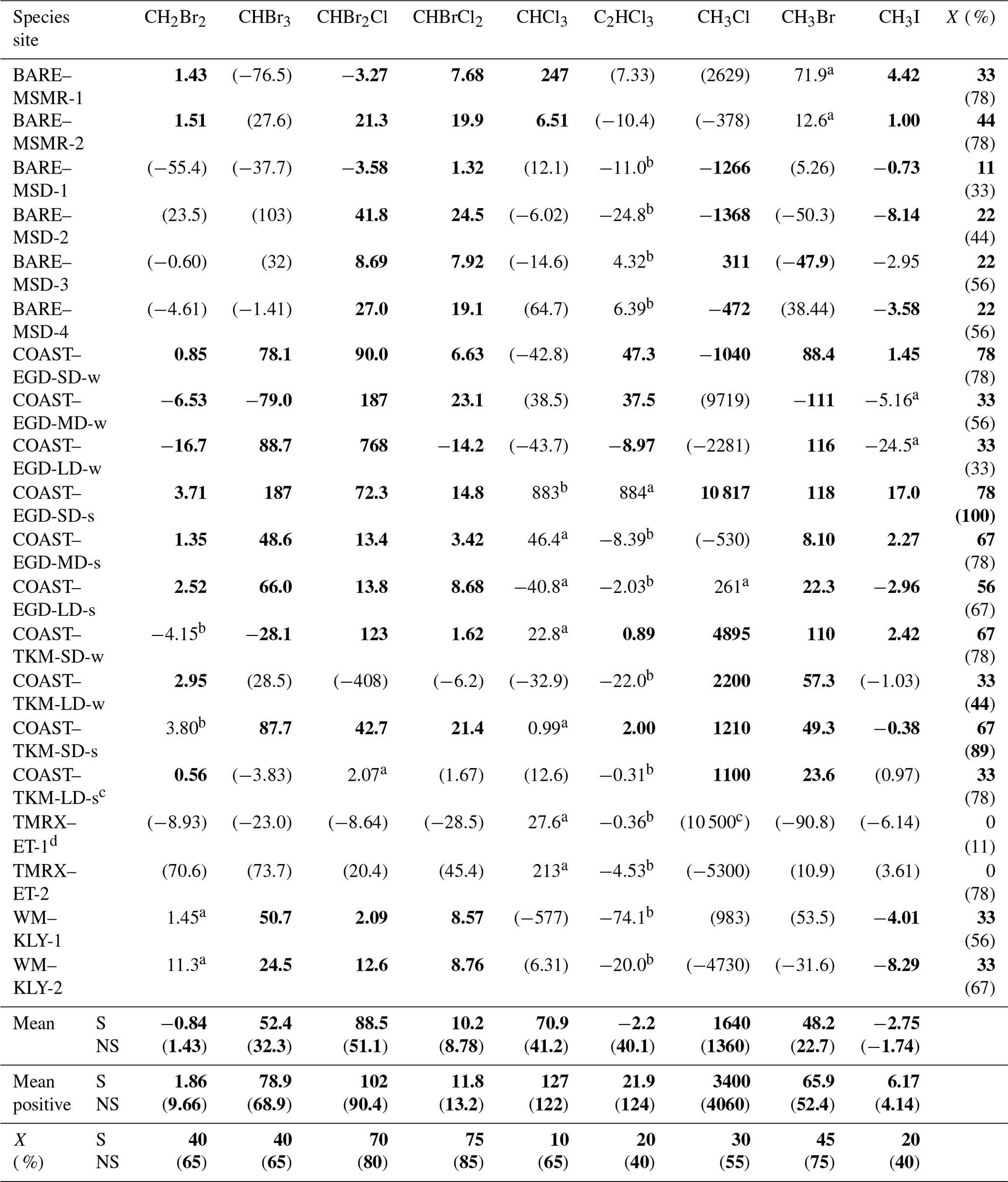
a 0.05 < p < 0.1 for a measurement site as net source or sink for a specific species. b 0.1 < p < 0.15 for a measurement site as a net source or sink for a specific species; S and NS indicate p < 0.05 and p > 0.05, respectively. c Flux calculation excludes one measurement for all VHOCs (see Sect. 2.1.2). d Flux calculation excludes one CH3Cl sample (see Sect. 2.1.2).
Table 2 presents the measured fluxes of all VHOCs studied alongside the corresponding statistical significance for a specific species' emission or depletion to a specific site. Note that considering the similar characteristics of the two SD sites, and of the two BARE sites, we assumed there to be a common emission source from the two sites, in both cases, in evaluating the statistical significance for these sites as a net source or net sink for the studied species. Considering the small number of measurements at each site, the table classifies the statistical significance of the fluxes' negative or positive values at a specific site into four different categories. While p values < 0.05 are used here to indicate statistical significance, p values of < 0.1 and < 0.15 are also indicated when present.

Figure 3VHOC fluxes at the different measurement sites. Fluxes associated with p values < 0.05 are marked by colored circles to indicate measurements during spring, winter and summer, with solid colored circles indicating measurements up to 3 d after a rain event in spring (Spring-prec.), up to 6 d after a rain event in winter (Winter-prec.) and in the evening in summer (Summer-eve.). Gray and black shapes indicate fluxes associated with no clear statistical significance (p > 0.1 and 0.05 < p < 0.1, respectively). At the center of each graph, the small black circles and error bars represent the average and standard error of the mean (SEM), respectively, for each measurement site. Dashed lines represent zero flux. In each box, the numbers indicate the mean flux and SEM (in parentheses) for each site and species. Additional information is provided for measurement conditions (Tables 1 and S6), measurement abbreviations (Table 1) and statistical analysis (Table 2). * Calculation of CH3Cl flux mean and SEM excludes one sample at TMRX–ET-1 (see Sect. 2.1.2). Calculation of mean flux and SEM excludes one sampling canister at COAST–TKM-LD (see Sect. 2.1.2).
Figure 3 presents the measured fluxes of all VHOCs studied individually, for statistically significant and non-significant fluxes emitted or depleted to a specific site. Non-significant fluxes are marked with black and gray for 0.05 < p < 0.1 and p > 0.1, respectively. It can be seen that for all species, at least one of the six studied areas could be classified as a net source, with slightly fewer sites being statistically significant net sources for CHCl3, C2HCl3 and CH3I. Note that as explained above, C2HCl3 was found to be affected by anthropogenic emission, which could explain the relatively less-frequent identified emissions for this species. Figure 3 clearly demonstrates that the COAST sites, and particularly the SD sites, are associated with the highest number of VHOCs with positive flux. These sites were also found to be a source for CHCl3, C2HCl3 and CH3I. Figure 3 does not indicate elevated VHOC emissions from the vegetated sites (WM–KLY and TMRX–ET) compared to the BARE sites.
The flux magnitudes for CHBr3 and CH2Br2 were greater than for most reported emissions in the MBL (e.g., CHBr3, 25.2–62.88 nmol m−2 d−1 for the Mauritanian upwelling – Quack et al., 2007; CH2Br2, 0.14–0.29 nmol m−2 d−1 for the New Hampshire coast – Zhou et al., 2008) but were smaller than the corresponding average fluxes estimated by Butler et al. (2007) for global coastal areas (∼220 and 110 nmol m−2 d−1, respectively) and than the average flux from the New Hampshire coast as reported by Zhou et al. (2005; and 113±130 nmol m−2 d−1, respectively).
Relatively high positive CHCl3 fluxes were measured for BARE–MSMR (247 nmol m−2 d−1), TMRX–ET-2 (213 nmol m−2 d−1) and COAST–EGD-SD-s (883 nmol m−2 d−1), although the latter two sites were not identified as a net source for CHCl3 (Table 2). For comparison, the emission from BARE–MSMR-1 was similar to the maximum emission found for tundra peat by Rhew et al. (2008), whereas the average emissions from COAST–EGD-SD-s and TMRX–ET-2 were comparable to those from temperate peatlands (∼496 nmol m−2 d−1 as measured by Dimmer et al., 2001). Whereas emissions for COAST–EGD-SD-s and TMRX–ET-2 might have been affected by seawater and vegetation, respectively, the emission for BARE–MSMR can be completely attributed to soil. The latter emission flux in BARE–MSMR was higher than the maximum emission rate in arctic and subarctic soils (∼115 nmol m−2 d−1) reported by Albers et al. (2017).
All investigated site types, except for the natural vegetation (TMRX–ET), were identified as net sources for CHBr2Cl and CHBrCl2 (Fig. 3). The mixing ratios of CHBr2Cl and CHBrCl2 were higher by factors of ∼4–14 and ∼5–11, respectively, than the average reported values for the MBL and were also higher than the mixing ratios measured in nearby coastal areas, except for the extremely high CHBr2Cl mixing ratios attributed to emission from a rock pool at Gran Canaria (ranging from 19 to 130 ppt; Ekdahl et al., 1998). For example, Brinckmann et al. (2012) found mean mixing ratios for CHBr2Cl and CHBrCl2 in coastal areas of the Sylt Islands (North Sea) of up to 0.2 and 0.1 ppt, respectively, while Mohd Nadzir et al. (2014) found CHBr2Cl and CHBrCl2 mixing ratios of 0.07–0.15 and 0.15–0.22 ppt, respectively, in the tropics. The measured CHBr2Cl fluxes for the Dead Sea were also higher than the reported value of 0.8 nmol m−2 d−1 (range of −1.2–10.8 nmol m−2 d−1) at coastal areas sampled during the Gulf of Mexico and East Coast Carbon Cruise (GOMECC; Liu et al., 2011). Typically, the net CHBrCl2 flux at the Dead Sea was significantly higher than corresponding fluxes from arctic and subarctic soils, as recently reported by Albers et al. (2017), ranging from 0.03–5.27 nmol m−2 d−1.
COAST–TKM and COAST–EGD-SD were found to be the only net source sites for CH3Cl. The highest positive fluxes were measured at COAST–EGD-SD and COAST–TKM-SD, with maximum net fluxes of ∼10 800 and 4900 nmol m−2 d−1, respectively. These fluxes are comparable in magnitude to those reported for several terrestrial sources, such as tropical forests (∼4520 nmol m−2 d−1), by Gebhardt et al. (2008) or by Yokouchi et al. (2002) and for other tropical or subtropical vegetation (Yokouchi et al., 2007), and they are higher than emissions from dryland ecosystems, including shortgrass steppe or shrublands (Teh et al., 2008). In some cases, the measured fluxes were higher than average emissions from salt marshes (e.g., ∼7300 nmol m−2 d−1; Deventer et al., 2018) but significantly smaller than the maximum fluxes from salt marshes (e.g., 570 000 nmol m−2 d−1; Rhew et al., 2000).
Both COAST–TKM and COAST–EGD sites were identified as net sources, while with less statistical significance (p < 0.1), BARE–MSMR was also identified as a net source of CH3Br (Table 2). In contrast to CH3Cl, emissions of CH3Br at the Dead Sea were significantly lower than the average reported emissions from marshes (e.g., ∼600 nmol m−2 d−1; Deventer et al., 2018). The fluxes measured at the Dead Sea were also lower than the reported emission from a coastal beach on a Japanese archipelago island (∼53 000 nmol m−2 d−1) but higher, in most cases, than in other dryland ecosystems (see Rhew et al., 2001).
Similar to CH3Br and CH3Cl, for CH3I, COAST–TKM and COAST–EGD, and particularly the SD sites, were identified as net sources (Table 2). BARE–MSMR was also identified as a net source for CH3I. Positive measured net fluxes of this compound were in most cases comparable to other reported fluxes over soil and vegetation. For example, Sive et al. (2007) reported a CH3I flux of ∼18.7 nmol m−2 d−1 over soil and vegetation at TF and a somewhat lower emission (∼12.6 nmol m−2 d−1) in Duke Forest, NC, USA. While the elevated flux at COAST–EGD-SD-s (17.0 nmol m−2 d−1) could potentially have been affected by flow of the sampled air over the seawater, the positive net fluxes at BARE–MSMR (1.00 and 4.42 nmol m−2 d−1) indicate significant emission from bare soil at the Dead Sea. The positive fluxes measured at BARE–MSMR were similar to the measured soil-emission fluxes of CH3I reported by Sive et al. (2007) at Duke Forest, averaging ∼0.27 nmol m−2 d−1 (range of ∼0.11–4.1 nmol m−2 d−1).
Only COAST–EGD and COAST-TKM-SD sites were found to be statistically significant sources (p < 0.05, see Table 2) for C2HCl3, suggesting that the elevated mixing ratios for this species in the Dead Sea area result mostly from local anthropogenic emissions. This possibility is supported by the high correlations with C2Cl4 (Table S5). Emissions from a more distant natural source, such as the Mediterranean Sea or Red Sea, are unlikely given their large distance (∼90 and ∼160 km, respectively).

Figure 4Bar graphs of VHOC fluxes from the different site types, organized by relative orientation to the Dead Sea and with visual indicators of surface cover type. Colored bars represent measured fluxes associated with p values < 0.05. Gray and black bars indicate fluxes associated with no clear statistical significance (black for 0.05 < p < 0.1 and gray for p > 0.1). Circles with drop lines are used to mark fluxes with relatively low values. Different colors refer to different VHOCs as indicated at the top of the figure. The different site types are indicated in the legend. S, M and L indicate short, medium and long distance of the measurement site from the seawater for the coastal sites (SD, MD and LD, respectively; see Sect. 2.1.1). See Table 1 for measurement sites and measurement abbreviations. * CH3Cl flux calculation excludes one sample in TMRX–ET-1 (see Sect. 2.1.2). Flux calculation excludes one sampling canister in COAST–TKM-LD (see Sect. 2.1.2).
3.2 Factors controlling VHOC flux
3.2.1 Seasonal, meteorological and spatial effects
The results presented in Sect. 3.1 showed elevated mixing ratios and net fluxes for all investigated VHOCs, with relatively less-frequent positive fluxes for CH3I, CHCl3 and C2HCl3. For all of the investigated VHOCs, a positive flux was measured for at least one of the two bare soil sites, BARE–MSMR and BARE–MSD, which are located a few kilometers from the Dead Sea water. For several VHOCs (CH2Br2, CHBr2Cl, CHBrCl2 and CHCl3), at least one of these sites was identified as a significant net source (p < 0.05, Table 2). Additional measurements are required to determine whether the other VHOCs are also emitted from these bare soil sites. Note that for all VHOCs except C2HCl3 and CH3Cl, measured mixing ratios were highest over at least one of these bare soil sites (Table S3). Figure 4 further provides the spatial distribution of the investigated VHOCs at the various sites. Elevated positive fluxes are seen at the coastal sites, with a general tendency toward higher positive net fluxes closer to the seashore. Figure 4 also demonstrates relatively high positive fluxes for the natural vegetation in TMRX–ET, higher than for WM–KLY. However, additional measurements are required to decipher whether this site can be classified as a statistically significant source for VHOCs (see Table 2).
No clear impact of meteorological conditions on the measured net flux rates or mixing ratios was observed. We could not identify any clear association between flux magnitude and any parameter, including solar radiation intensity, measurement time, temperature and daytime relative humidity.
Our findings on the effects of season and distance from the sea on the measured fluxes are presented in Fig. 3, which shows the measured fluxes for spring and winter and for different distances from the sea at COAST–EGD and COAST–TKM. Differences in VHOC emissions between winter and spring may arise from the generally much higher temperature and lower precipitation during the latter; further considering the high evaporation rate in this area, the soil water content is expected to be generally lower in spring compared to winter (Sect. 2.1.1; see also Table S6). Figure 3 suggests that there were no clear differences in VHOC fluxes between spring and winter, as supported by statistical analysis, except for CH3I and CH2Br2, for which fluxes were higher in the spring, with moderate statistical significance (0.05 < p < 0.1).
No clear impact of distance from the seawater on the measured net fluxes could be detected, including in cases where a significant fraction of the footprint included the seawater, such as for COAST–EGD-SD-w and COAST–EGD-SD-s. However, owing to variations in soil properties, the emissions near the seawater tended to be more frequent and more intense (see Sect. 3.2.2, 3.2.3).
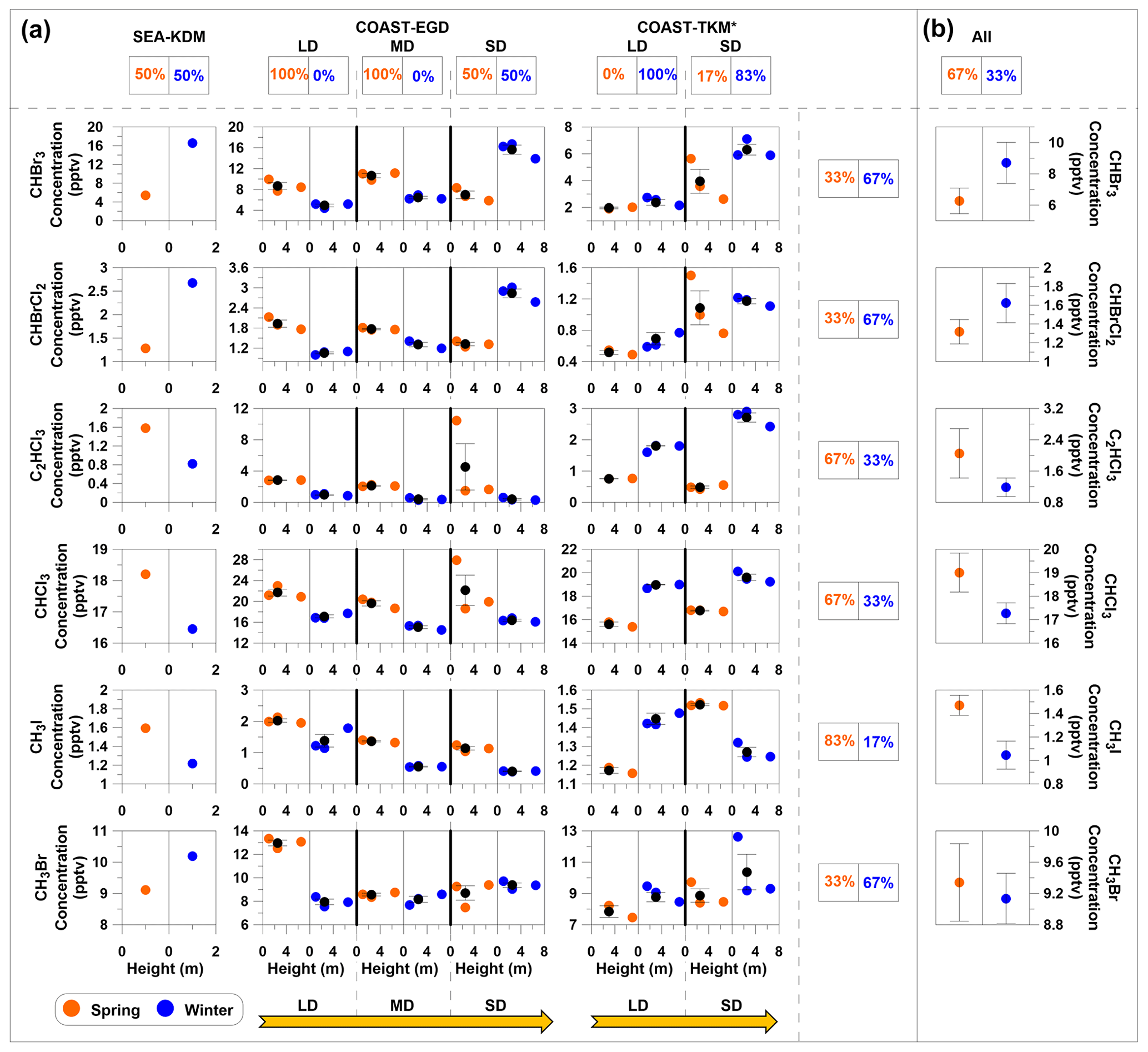
Figure 5Seasonal and spatial influences on measured mixing ratios of VHOCs for coastal sites only. (a) Measured VHOC mixing ratios are presented vs. vertical height above surface level, separately for winter (blue) and spring (orange). Black filled circles and error bars represent average and standard error of the mean (SEM), respectively. LD, MD and SD indicate long, medium and short distance from the seawater, respectively, while SEA–KDM is located at the seawater (see Sect. 2.1.1). Values above and to the right of the figure indicate the percentages of higher average mixing ratios in spring (left box) or winter (right box) individually, for each site (SEA–KDM, COAST–TKM and EGD sites) and for each specific species. (b) For each species, the average mixing ratios over all sites (SEA–KDM, COAST–EGD and COAST–TKM) are presented (All), and the corresponding percentage of higher average mixing ratios in spring and in winter are also presented. See Table 1 for measurement site abbreviations. Species with no observed difference between seasons were excluded (see Fig. S1 in the Supplement for complete information); y axes for sites in the same coastal area (COAST–TKM or COAST–EGD) are evenly scaled. * Measurements exclude one sampling canister at COAST–TKM-LD (see Sect. 2.1.2).
Figure 5 compares the mixing ratios of the measured VHOCs at different distances from the seawater individually for winter and spring. Note that differences in sampling heights at different sites can lead to a biased comparison between mixing ratios at different sites; nevertheless, in most cases, differences across measurement sites were larger than across vertical heights. No clear impact of season or distance from the seawater on the mixing ratios can be discerned in this figure, also based on the sampling over SEA–KDM, which directly represents air masses over the seawater (Sect. 2.1.1). Nevertheless, further investigation, using direct flux measurements over the Dead Sea water, is needed to study the potential emission of VHOCs from this water body. While no clear impact of season on mixing ratios was observed, for most sites, differences between two measurement sets resulted in consistent differences in mixing ratios such that one measurement set resulted in higher mixing ratios for all or most species than the other. This suggests that other factors play a significant role in emission rates of all or most VHOCs in the studied area. Only the CH3I results indicated moderate statistical significance (0.05 < p < 0.1) for higher mixing ratios in the spring vs. winter, in agreement with seasonal trends for its flux, as discussed above.
3.2.2 Impact of specific site characteristics and ambient conditions
The formation of VHOCs requires a chemical interaction between OM and halides, induced by biogeochemical, biochemical or macrobiotic processes (Kotte et al., 2012; Breider and Albers, 2015). Despite the extreme salinity, biotic activity was detected in both the water and the soil of the Dead Sea (see Sect. 1), demonstrating that biotic activity can potentially contribute to VHOC emission in this area. Previous studies on emission of VHOCs from soil and sediments revealed that OM content and type, halide ion concentrations, pH, and the presence of an oxidizing agent (most frequently referred to as Fe(III)) also play important roles in the emission rate of VHOCs (see Kotte et al., 2012).
Table 3Soil properties – OM, soil water content (SWC), I, Br, Cl and Fe fraction of dry weight and pH. Analyses were performed for a single mixture of samples at each site. See Table 1 for measurement site abbreviations.
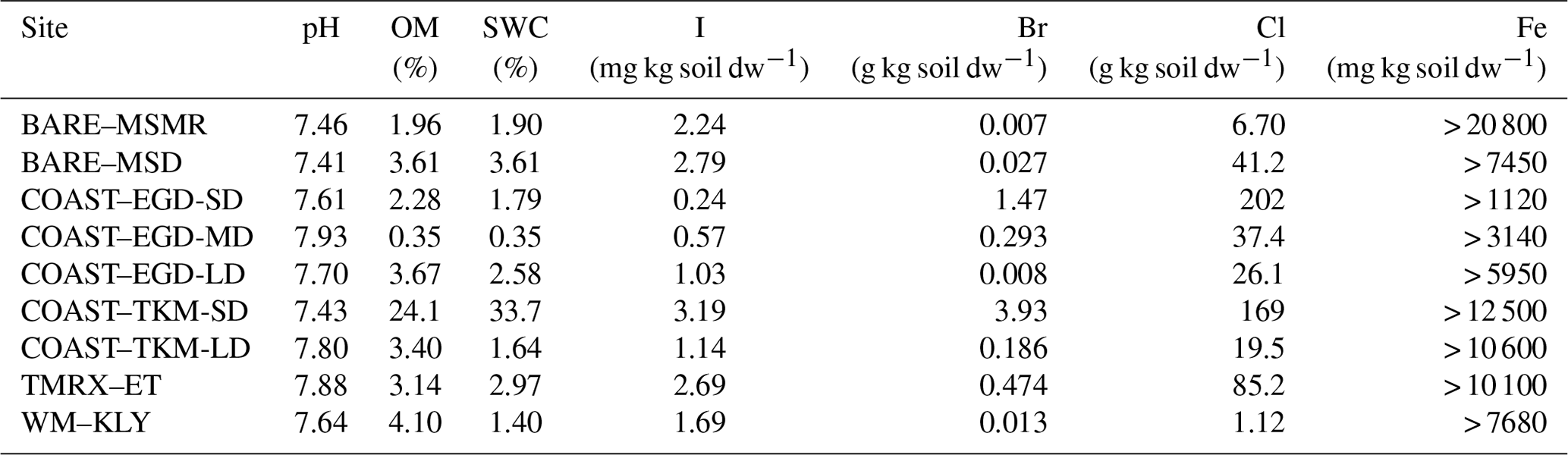
Table 3 provides a basic representation of the soil composition parameters. The results presented in Table 3 show substantial enrichment of Cl and Br in the sites closest to the seawater (COAST–EGD-SD and COAST–TKM-SD) and lower concentrations at greater distances from the seawater. For comparison, both Br and Cl concentrations were generally much higher than those reported by Kotte et al. (2012) for various saline soils and sediments (0.12–0.32 and 6.1–120 g kg−1, respectively) but lower for Br at BARE–MSMR and BARE–MSD and for both Cl and Br at WM–KLY. No enrichment of I in the soil samples was observed (e.g., Keppler et al., 2000; Kotte et al., 2012). The OM content of the samples was generally higher than would be expected in desert soil. For comparison, forest floors typically contain 1 %–5 % OM (Osman, 2013). Detection of VHOC emissions from the soil is, in some cases, associated with higher soil OM (e.g., Albers et al., 2017; Keppler et al., 2000) and, in some cases, with lower soil OM (e.g., Kotte et al., 2012; Huber et al., 2009) than that reported here. Table 3 provides only a lower limit of the total Fe, rather than Fe(III), in the samples. Note, however, that soil Fe content similar to that reported here as a low-limit value corresponds with that associated with the finding of small amounts of VHOC emissions, while the emission rates become saturated when enrichment with Fe(III) is relatively minor (Keppler et al., 2000). Saturation at relatively low soil Fe concentrations was also reported by Huber et al. (2009). Hence, variations in Fe across different sites may play a minor role in affecting emission rates.
While the number of samples collected at each site was limited, Table 2 and Fig. 4 indicate elevated positive fluxes for the SD sites, and to some extent also at COAST–EGD-MD, with respect to both statistically significant and non-statistically significant positive fluxes. Moreover, for both COAST–EGD and COAST–TKM, during both spring and winter, the occurrence of positive fluxes was correlated with proximity to seawater (i.e., COAST–EGD-SD > COAST–EGD-MD > COAST–EGD-LD and COAST–TKM-SD > COAST–TKM-LD). All of these COAST sites contain mixtures of soil and salt-deposited structures (see Sect. 2.1.1), and Table 3 indicates that soil concentrations of both Br and Cl correlated with proximity to seawater at both COAST–EGD and COAST–TKM. The concentration of I in the soil showed a similar trend only at the COAST–TKM sites (see Table 3). The association between the magnitude and incidence of the positive net flux and soil halide concentrations points to an increase in VHOC emission with salinity, even under the hypersaline conditions of the Dead Sea area. This interpretation is supported by the fact that whereas for COAST–TKM-SD, both soil water and OM content were relatively high, for COAST–EGD-SD, no other measured parameter which could limit the emission of VHOCs, except for the soil halide concentration, was higher than for both COAST–EGD-MD and COAST–EGD-LD (Table 3). The fact that emission rates for COAST–TKM tended to be similar or lower in terms of incidence and magnitude compared to COAST–EGD (Table 2) suggests, in view of the apparently lower Fe content for the latter (Table 3), that the emission of VHOCs from these sites is not significantly limited by the availability of Fe(III) in the soil.
COAST–EGD-SD-s was associated with the highest incidence of both statistically significant and non-significant positive fluxes. Fluxes at COAST–EGD-SD-w were generally lower and with a smaller incidence of positive fluxes. Based on the wind direction, in both cases, the sampling footprint included both the seawater and a narrow strip of bare soil mixed with salty beds (estimated at about 60 % of the footprint) very close to the seawater. The main notable difference between the two measurement days was that precipitation occurred just before the COAST–EGD-SD-w measurement, whereas there was no precipitation event for several weeks prior to the COAST–EGD-SD-s measurement (Table 1). Rain events also occurred ∼1.5 and ∼2.5 d before BARE–MSD-1 and BARE–MSD-2 measurements, respectively. Note that the emission fluxes for BARE–MSD-1 were lower and more negative for most of the species than those for BARE–MSD-3 or BARE–MSD-4. In addition, the occurrence of positive net fluxes tended to increase according to the order BARE–MSD-1 < BARE–MSD-2 < BARE–MSD-3 (see Table 2). The analyses for both COAST–EGD and BARE–MSD suggest that increased soil water content caused by rain events can decrease the emission rates or enhance soil-uptake rates of certain VHOCs.
A reduction in net flux rates following rain events did not occur for all species and was not clearly consistent across the BARE–MSD and COAST–EGD-SD sites. Thus, further research on the effects of rain on the various VHOCs and ambient conditions is required. Nevertheless, the analyses clearly demonstrate that strong emission rates do not depend on rain occurrence, in agreement with findings by Kotte et al. (2012). The lower emission fluxes following the rain event may be attributable to the low infiltration rate of VHOCs into the soil, to salt dilution and washout, or both.
Our measurements suggested an elevated contribution of natural vegetation to some of the investigated VHOCs (Fig. 4), but with no statistical significance for this site being a source of any of the investigated VHOCs (Table 2). This might reflect the fact that only a few measurements are available for this site. No clear contribution of the agricultural vegetation to the emission fluxes was found in this study.
3.2.3 Factors controlling the flux of specific VHOCs
Trihalomethanes. Differently from previous studies, brominated VHOCs had relatively higher overall incidence of positive fluxes than chlorinated VHOCs (Table 2). The overall average net flux of trihalomethanes decreased according to CHBr2Cl > CHCl3 > CHBr3 > CHBrCl2, while CHCl3 showed the lowest incidence of positive and highest mean positive fluxes among all trihalomethanes.
Natural emission of trihalomethanes from soil has been shown to occur without microbial activity, induced via oxidation of OM by an electron acceptor such as Fe(III) (Huber et al., 2009) or via hydrolysis of trihaloacetyl compounds (Albers et al., 2017). The soils studied by Albers et al. (2017) were significantly richer in OM than the soils at the Dead Sea, except for COAST–TKM-SD. Hence, the apparently higher emission from the Dead Sea soil may indicate either a different mechanism leading to the release of trihalomethanes from the soil or only a weak dependency on availability of soil OM. The latter explanation may be supported by the fact that Albers et al. (2017) did not find any correlation between the CHCl3 emission rate and organic Cl in the soil. Furthermore, our study points to higher emission rates and incidence of VHOCs, and generally also of trihalomethanes, closer to the seawater (COAST–EGD and COAST–TKM sites), which suggests higher sensitivity to soil halide content than OM (Sect. 3.2.2).
While trihalomethane formation via OM oxidation has been reported to occur more rapidly at low pH, and specifically at pH < ∼3.5 (Huber et al., 2009; Ruecker et al., 2014), its formation via hydrolysis of trihaloacetyl is expected to occur more rapidly at the relatively high pH ≥7 (Hoekstra et al., 1998; Albers et al., 2017). Yet according to Ruecker et al. (2014), in hypersaline sediments, the formation of VHOCs via OM oxidation involving Fe(III) can occur at pH > 8 for biotic processes. Therefore, given the relatively high pH (∼7.4–7.9; Table 3) at the SD sites as well as the BARE and WM–KLY sites, the high trihalomethane-emission rates from both bare and agricultural field sites support the work by Albers et al. (2017) concerning the emission of trihalomethanes from the soil following trihaloacetyl hydrolysis.
Albers et al. (2017) showed that their proposed mechanism supports the emission of CHCl3 and CHBrCl2 from soil and suggested that additional halomethanes with a higher number of Br atoms can be expected to be emitted via this mechanism but at much lower rates. Hence, the elevated net fluxes for CHBr2Cl and CHBr3 at the Dead Sea (Table 2) could occur either because of the markedly higher composition of Br in the Dead Sea soil (see Table 3) or because another mechanism is also playing a role in the emission; note that agriculture could potentially be a source for the emission of CHBr2Cl and CHBr3 for WM–KLY but not for the other sites (Sect. 2.1.1). The finding of Hoekstra et al. (1998) that Br enrichment mainly enhances the emission of CHBr3 and CHBr2Cl, rather than that of CHBrCl2, supports the former possibility, namely, relatively elevated emission of CHBr2Cl and CHBr3 due to higher Br content in the soil. While both Cl and Br soil contents are relatively high for both COAST SD sites and COAST–EGD-MD, where emission of brominated trihalomethanes was higher than that of chlorinated trihalomethanes (see Table 2), a remarkably high Br∕Cl value (1:43) relative to other sites was found at COAST–TKM-SD. Table 2 does not indicate a clear difference in the flux magnitude of the brominated compared to chlorinated trihalomethanes for this site, suggesting that the main reason for the relatively elevated brominated trihaloethanes at the SD sites and COAST–EGD-MD is the high Br content rather than the Br∕Cl ratio.
The relatively elevated net flux of brominated trihalomethanes from BARE and WM–KLY indicates that relatively high rates of emission of these species can also occur from soils that are much less rich in Br than the SD sites and the COAST–EGD-MD site (see Tables 2, 3). Yet the emission rates of CHBrCl2 at the Dead Sea were generally higher than those observed by Albers et al. (2017), probably reflecting the higher soil Cl content at the Dead Sea.
Methyl halides. A relatively high incidence of negative fluxes was observed for CH3Br, and more statistically significantly so for CH3Cl and CH3I, implying high rates of both emission and deposition, at least for the latter two, in the studied area (Table 2). The average positive flux of CH3Cl was the highest of all VHOCs investigated, indicating strong emission and deposition for this species at the Dead Sea. Several studies have indicated that soil tends to act as a sink for CH3Cl (Rhew et al., 2003). The relatively high positive net fluxes of CH3Cl and CH3Br at WM–KLY-1 (983 and 53.5 nmol m−2 d−1, respectively) may point to emission of this species from the local agricultural field, in agreement with previous studies (Sect. 1), potentially by microbially induced or fungus-induced emission (Moore et al., 2005; Watling and Harper, 1998), but this should be further investigated, considering the lack of statistical significance.
Positive net fluxes for CH3I were not significantly higher than those obtained in previous studies (Sect. 3.1), a finding that might be attributed to the small concentration of I in the soil relative to those of the other halides. At Duke Forest, Sive et al. (2007) observed a soil-emission CH3I flux of ∼0.27 nmol m−2 d−1 on average (ranging from ∼0.11 to 0.31 nmol m−2 d−1) under precipitation conditions in June and higher emission rates (0.8 and 4.1 nmol m−2 d−1) under warmer and dryer conditions in September. In agreement with those findings, although in general our analyses did not indicate clear seasonal effects, we found that in all cases, net CH3I fluxes were higher in spring than in winter, except for COAST–TKM-SD (Fig. 3). As discussed in Sect. 3.2.1, the mixing ratios of CH3I also tended to be higher in magnitude in spring compared to winter, with moderate statistical significance (0.05 < p < 0.1 in both cases; Figs. 3, 5).
Relatively high fluxes of CH3Cl and CH3Br and, to a lesser extent, of CH3I were observed at the COAST–TKM and COAST–EGD sites, particularly from the sites closest to the seawater (Fig. 4). According to Keppler et al. (2000), the presence of Fe(III), OM and halide ions is basically enough to result in emission of methyl halides from both soil and sediments by a natural abiotic process (Sect. 1). The strong emission of methyl halide from the COAST–TKM and COAST–EGD sites indicates that these species can be emitted at high rates from saline soil that is not rich in OM. The strongest emissions occurred from COAST–TKM-SD and COAST–EGD-SD, which may indicate high sensitivity of methyl halide emission to soil OM and/or halide content (see Table 3). The fact that the emission of methyl halides, particularly CH3Br and CH3Cl, from COAST–TKM-SD, where soil OM is substantially higher than at all other investigated sites, was not higher than the emission from COAST–EGD-SD-s may indicate that emission of methyl halides was not sensitive to soil OM in our study. Note that the lower fluxes for EGD-SD-w compared to EGD-SD-s can be associated to a prior rain event for the former (Sect. 3.2.2).
In controlled experiments to study emissions of the three methyl halides from soil, Keppler et al. (2000) found a decrease in the efficiency of methyl halide emission according to CH3I > CH3Br > CH3Cl (; mole fractions). We estimated the emission efficiencies of the different methyl halides based on the ratio between their fluxes and the concentrations of halide in the soil. To maintain consistency with the calculations of Keppler et al. (2000), our calculation was also based on mole fractions and took into account only positive fluxes, on the assumption that they are closer in magnitude to emission. This corresponded with measured soil halide concentration proportions for of , and the evaluated emission efficiency proportions were for CH3I, CH3Br and CH3Cl, respectively, when two outliers were excluded from the calculations. These calculations confirmed the increasing efficiency of methyl halide emission following CH3Cl < CH3Br < CH3I, in agreement with Keppler et al. (2000), suggesting that at least the methylation and emission of CH3Br and CH3Cl in our study were controlled by abiotic mechanisms similar to those reported by Keppler et al. (2000). The apparently higher relative efficiency of CH3I emission may indicate emissions of CH3I via other mechanisms in the studied area, as discussed in Sect. 3.3. It should be noted, however, that the fluxes that we used for the methyl halide emission efficiencies were based on measured net flux rather than measured emission flux. This might also explain the inconsistency between the relative CH3I-emission efficiency calculated by Keppler et al. (2000) and by us.
C2 HCl3. C2HCl3 had the second-lowest incidence of positive fluxes, with statistically significant (p < 0.05) positive fluxes only from the COAST SD sites and COAST–EGD-MD (Table 2). These sites are mixtures of salt beds and deposits with salty soil, and therefore the elevated emissions of C2HCl3 at these sites appear to support previous evidence for the emission of this gas by halobacteria from salt lakes, as reported by Weissflog et al. (2005). Additional chlorinated VHOCs, including CHCl3 and CH3Cl, also demonstrated increased emission from this site, in line with the findings of Weissflog et al. (2005). Note that the net measured fluxes for most of the VHOCs investigated at the COAST–EGD-SD-w site were smaller than those at COAST–EGD-SD-s, as discussed in Sect. 3.2.2.
CH2Br2. CH2Br2 showed positive fluxes from all site types, with a positive average net flux from most sites (see Fig. 3), but its fluxes over the vegetated and agricultural sites were not statistically significant. Correlation of CH2Br2 with trihalomethanes will be discussed in Sect. 3.3.
Table 4Correlations between the mixing ratios of VHOCs. Shown is the Pearson correlation coefficient (r) between each VHOC pair for the measured mixing ratio, when calculated over all sites excluding SEA–KDM (NO-KDM), all sites (ALL), bare soil sites (BARE), coastal sites (COAST), short distance from the sea at the coastal sites (SD) and the vegetated sites (VEG). Correlations were calculated for mean mixing ratios at each site. The p value for r being significantly different from zero is indicated based on one-sample t test, in four categories. For values in bold, p<0.05. For values in parentheses, p > 0.15. ap ≪0.1. bp < 0.15.
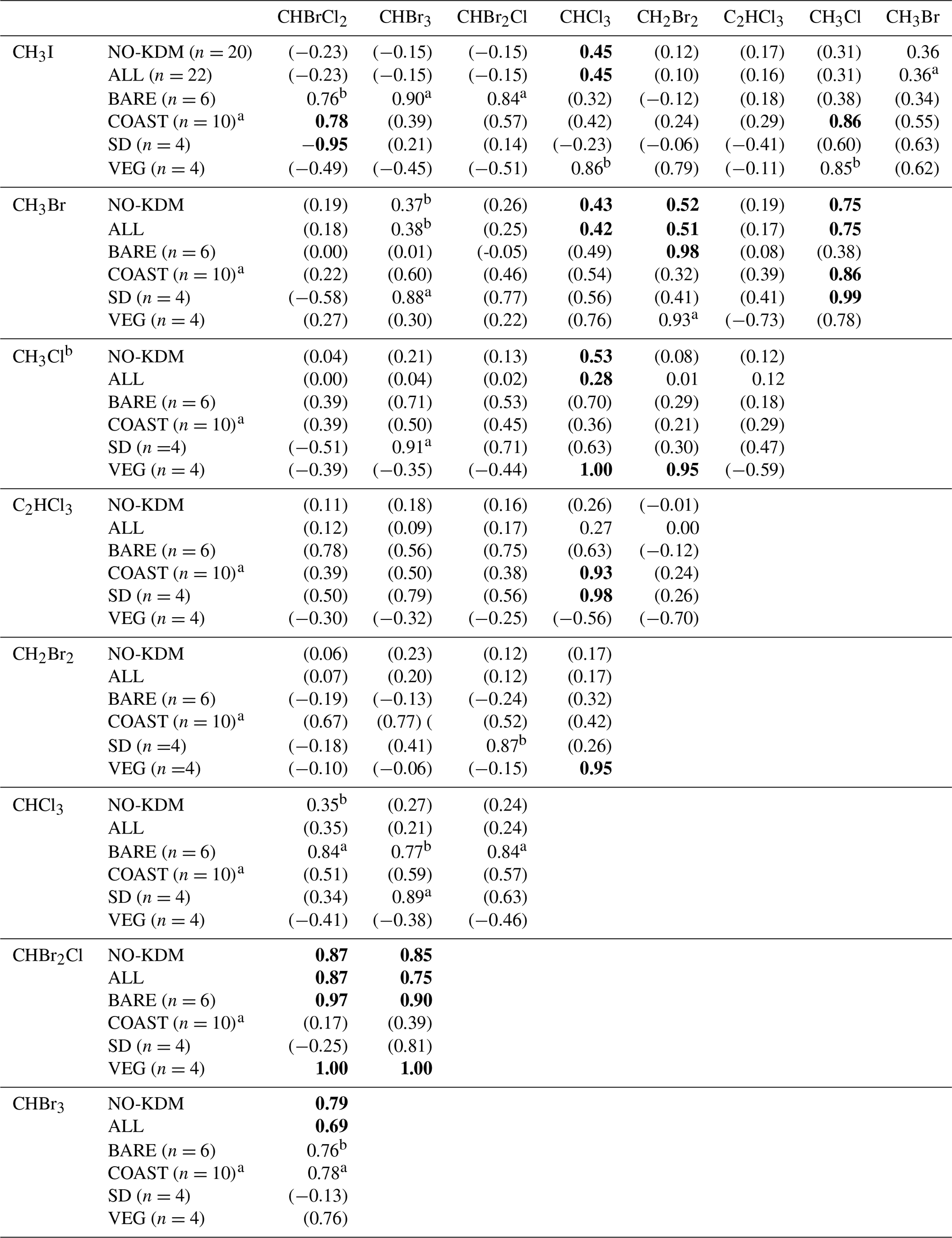
a Correlation calculation for COAST–TKM-LD excluded one sampling canister (see Sect. 2.1.2). b Correlation calculation for CH3Cl excluded one sample for TMRX–ET-1 (see Sect. 2.1.2).
3.3 Flux and mixing ratio correlations between VHOCs
Table 4 presents the Pearson correlation coefficients (r) between the measured mixing ratios of VHOCs at the Dead Sea, separately for all sites and for the terrestrial sites only as well as separately for BARE, COAST, and the natural vegetation and agricultural field sites (VEG). For COAST, r is also presented individually for the two sites which were closest to the seawater (SD). The correlations' significance levels are also indicated. In most cases, the correlations between species over all terrestrial sites were low but were substantially higher for the brominated trihalomethanes (CHBr3–CHBrCl2 – r=0.79; CHBr2Cl–CHBrCl2 – r=0.87; CHBr2Cl–CHBr3 – r=0.85), supporting a common source mechanism for these species. High correlations between these three trihalomethanes can be attributed to high correlations at the BARE and VEG sites. Relatively high correlations were also obtained, although to a lesser extent, between methyl halides, particularly between CH3Cl and CH3Br (r=0.75), which can be attributed to correlations at the COAST sites, particularly at the SD sites. For COAST, and particularly for SD, a high correlation was observed between C2HCl3 and CHCl3. Correlations were in most cases either similar or smaller when we included measurements from the seawater site SEA–KDM, which may reinforce the notion that emission from the seawater does not contribute significantly to VHOC mixing ratios in the area of the Dead Sea.
Table 5Correlations between the measured net fluxes of VHOCs. The table records the Pearson correlation coefficient (r) for the measured net flux between each VHOC pair, calculated over all sites except SEA–KDM (ALL), bare soil sites (BARE), coastal sites (COAST), short distance from the sea at the coastal sites (SD) and the vegetated sites (VEG). The p value for r, being significantly different from zero, is indicated based on t test in four categories. By default, for bolded values, p < 0.05. For values in parentheses, p > 0.15. ap < 0.1. bp < 0.15.
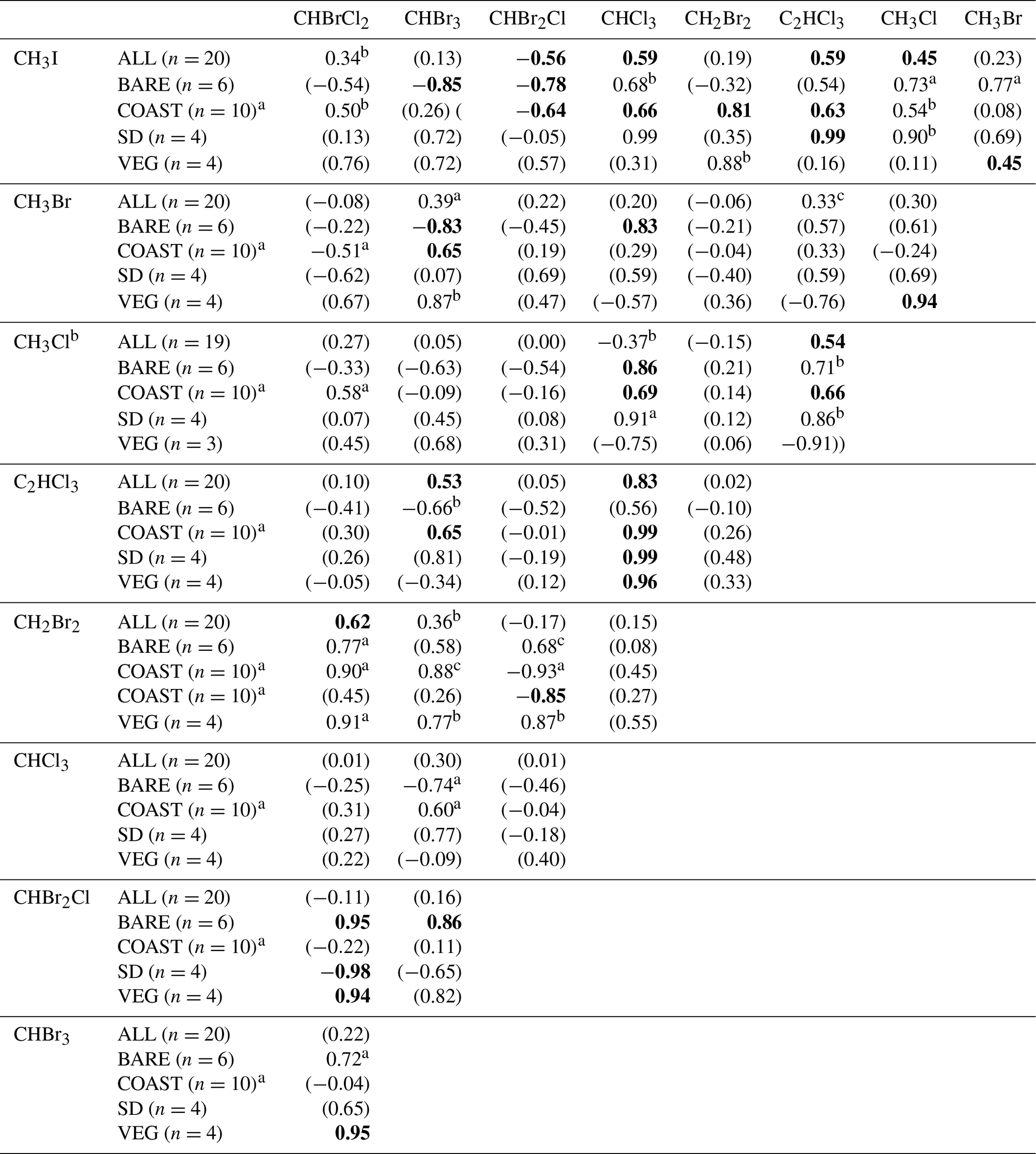
a Correlation calculations for COAST–TKM-LD excluded one sampling canister (see Sect. 2.1.2). b Correlation calculation for CH3Cl excluded one sample for TMRX–ET-1 (see Sect. 2.1.2).
Table 5 shows the correlations between the measured VHOC fluxes, separately for all sites (ALL), BARE sites, VEG sites, the TMRX–ET site and WM–KLY site, and COAST–TKM and COAST–EGD sites. For the latter two sites, correlations are also presented separately for the SD sites. Note that the table compares net flux rather than emission flux, and therefore the reported correlations are expected to be affected by both sinks and sources for the different VHOCs.
The results in Table 5 show moderate to high positive correlations in most cases when all sites are included in the calculation, whereas in many cases, the correlations were significantly higher when calculated for sites of the same type, suggesting common emission mechanisms or controls. High correlations were obtained for VEG between CHBrCl2, CHBr2Cl and CHBr3 (r≥0.94; p < 0.05), except for the correlation between CHBr2Cl and CHBr3 (r=0.82; p > 0.15). Note that these correlations can potentially be attributed to agricultural emission, considering that WM–KLY, but not TMRX–ET, was identified as a statistically significant source for the three trihalomethanes. At the BARE sites, high positive correlations between the fluxes of the three brominated trihalomethanes were observed which were all associated with p values < 0.05, except for a lower correlation between CHBr3 and CHBrCl2 (r=0.72; p < 0.1). Furthermore, high correlations between the mixing ratios of the three trihalomethanes were obtained for these two sites, although relatively low statistical significance was obtained for the correlation between CHBr3 and CHBrCl2 at these sites (see Table 4). This further supports the notion that the three brominated trihalomethanes are emitted via similar mechanisms or controls. Moderately low p values for the correlations between CH2Br2 and both CH2Br2Cl (p < 0.15) and CHBrCl2 (p < 0.1) at these sites further suggests common controls for CH2Br2 and the brominated trihalomethanes (see Table 5).
Correlation of CH2Br2 with CHBr2Cl at the SD sites was strongly negative (; p < 0.1), similar to the negative correlation between CHBr2Cl and the other brominated trihalomethanes, CHBrCl2 (; p < 0.05) and CHBr3 (; p > 0.15), at these sites. This, together with the fact that the measured fluxes of these three species were generally positive over the SD sites, suggests competitive emission between CHBr2Cl and CHBrCl2 and potentially also CHBr3, at least at the SD sites. This is supported by the analysis in Sect. 3.2.2 and 3.2.3, which demonstrated that the halide content of the soil appears to play a major role in controlling the emission rates of VHOCs under the studied conditions.
Table 5 also indicates overall low correlations between CHCl3 and all of the brominated trihalomethanes, mostly resulting from negative correlations at the BARE sites. The anticorrelation of CHCl3 with trihalomethanes increased in the order CHBrCl2 < CHBr2Cl < CHBr3. The incidence of the chlorinated trihalomethanes (CHCl3 and CHBrCl2), compared to the less chlorinated ones (CHBr3 and CHBr2Cl) also tended to be higher at the BARE sites compared to the other sites (Table 2). Hence, the negative correlation between CHCl3 and the brominated trihalomethanes at the bare soil sites may indicate competitive emission between the more chlorinated and more brominated trihalomethanes. The situation at the BARE sites resembles previous reports of predominant emission of CHCl3 at the expense of the more brominated species (e.g., Albers et al., 2017; Huber et al., 2009), particularly CHBr3 and CHBr2Cl, and was expected given the higher Cl / Br ratio at these sites (see Table 3). We should emphasize that even at the BARE sites, we observed relatively high positive fluxes of brominated trihalomethanes, particularly of CHBr2Cl and CHBrCl2, which would not generally be expected (Albers et al., 2017) and can be attributed to the relatively high Br enrichment in the soil.
Interestingly, in agreement with Table 4, Table 5 also shows relatively high correlations between CHCl3 and all methyl halides, particularly for the BARE sites (CH3I, r=0.68, p < 0.15; CH3Br, r=0.83, p < 0.05; CH3Cl, r < 0.86, p < 0.05) and SD sites (CH3I, r=0.99, p < 0.05; CH3Br, r=0.59, p > 0.15; CH3Cl, r=0.91, p < 0.1). Remarkably, a high correlation was found for CH3I with CHCl3 and C2HCl3 at the SD sites (r=0.99, p < 0.05 in both cases). Positive fluxes of the three species were observed at the SD sites in most cases, although with only moderate statistical significance for CHCl3 (Table 2). Weissflog et al. (2005) found that emission of C2HCl3, CHCl3 and other chlorinated VHOCs can occur from salt lakes via the activity of halobacteria in the presence of dissolved Fe (III) and crystallized NaCl. The strong correlations of CHCl3, C2HCl3 and CH3I at the SD sites, where statistically significant fluxes were frequently measured for these species, reinforce the co-located emissions of CHCl3 and C2HCl3 from salt lake sediments, as indicated by Weissflog et al. (2005), and suggest that CH3I can be emitted in a similar fashion. The fact that the relative emission efficiency of CH3I in our study was much higher than under the conditions used by Keppler et al. (2000) supports the possibility that mechanisms other than the abiotic emission pathway proposed by Keppler et al. (2000) influence the emission of CH3I at the Dead Sea (Sect. 3.2.3).
The results of this study demonstrate high emission rates of the investigated VHOCs in the Dead Sea region, corresponding with mixing ratios which, in most cases, are significantly higher than typical values in the MBL. Overall, our measurements indicate a generally elevated incidence of positive fluxes of brominated vs. chlorinated VHOCs compared to previous studies. The high incidence of the former can be attributed primarily to the relatively large amount of Br in the soil rather than the Br∕Cl ratio. We did not detect any clear effect of meteorological parameters, emission from the seawater or the season, other than – in agreement with Sive et al. (2007) – apparently higher emission of CH3I in spring vs. winter. Three of the investigated site types – bare soil, coast and agricultural field – were identified as being statistically significant (p < 0.05) sources for at least some of the investigated VHOCs. The fluxes, in general, were highly variable, showing changes between sampling periods, even for a specific species at a specific site. The coastal sites, particularly at a short distance from the sea (SD sites) where soil is mixed with salt deposits, were sources for all of the investigated VHOCs but not statistically significantly for CHCl3. Further from the coastal area, the bare soil sites were sources for CHBrCl2, CHBr2Cl, CHCl3 and apparently also for CH2Br2 and CH3I, and the agricultural vegetation site was a source for CHBr3, CHBr2Cl and CHBrCl2. Our measurements reinforce reports of CHCl3 and CHBrCl2 emission from bare soil but indicate that such emission can also occur under relatively low soil organic content. To the best of our knowledge, we report here for the first time strong emission of CHBr2Cl and emission of CH2Br2 from hypersaline bare soil, at least a few kilometers from the Dead Sea. We could not identify the contribution of either natural or agricultural vegetation to the emission of the investigated VHOCs.
The highest emissions from the SD sites were associated with maximum salinity and clearly showed an increased incidence of positive flux with proximity to the seawater, pointing to the sensitivity of VHOC emission rates to salinity, even under hypersaline conditions. The measurements did not indicate either increased or reduced emissions of VHOCs from the seawater itself. Emission of VHOCs has been shown to occur from dry soil under semiarid conditions during the summer, in agreement with the finding from other geographical locations that soil water does not seem to be a limiting factor in VHOC emission (Kotte et al., 2012). Rain events appeared to attenuate the emission rates of VHOCs at the Dead Sea. Measurements at a bare soil site suggested a decrease in VHOC emission rates for 1–3 d after a rain event.
Both flux and mixing ratio correlation analyses pointed to common formation and emission mechanisms for CHBr2Cl and CHBrCl2, in line with previous studies, for the agricultural watermelon-cultivation field and bare soil sites. These analyses further strongly suggest common formation and emission mechanisms for CHBr3 with these two trihalomethanes. Whereas Albers et al. (2017) suggested that CHBr3 and CHBr2Cl are emitted from soil only in relatively small amounts compared to CHCl3, our results indicated their high emission via common mechanisms with the other trihalomethanes. The overall average net flux of the trihalomethanes decreased according to CHBr2Cl > CHCl3 > CHBr3 > CHBrCl2, while CHCl3 showed the lowest incidence of positive fluxes among all trihalomethanes. The enhanced emission of brominated trihalomethanes probably reflects enrichment of the Dead Sea soil with Br, in line with findings by Hoekstra et al. (1998).
We identified the coastal sites as being a probable source for all methyl halides, whereas neither the agricultural field nor natural vegetation site was identified as net sink or net source for these species, except for the agricultural field being a net sink for CH3I. Our analysis demonstrated, however, much higher efficiencies of CH3I emission than of CH3Br and CH3Cl emissions as a function of halides in the soil, compared to those reported by Keppler et al. (2000), pointing to emission of CH3I via other mechanisms. The strong correlation between both fluxes and mixing ratios of CH3I, CHCl3 and C2HCl3, particularly at the SD sites, strongly suggests that the coastal area of the Dead Sea acts as an emission source for CHCl3, C2HCl3 and CH3I via similar mechanisms, although these sites were associated with only moderate statistical significance (p<0.1) as a net source for CHCl3. The emission of CHCl3 and C2HCl3 from these sites is in line with findings by Weissflog et al. (2005) of emission of various chlorinated VHOCs, including CHCl3 and C2HCl3, from salt lake sediments. Weissflog et al. (2005) reported that the emission of chlorinated VHOCs in their study was induced by microbial activity. Keppler et al. (2000) reported the involvement of an abiotic process in the formation of alkyl from soil and sediments, and the observed correlation between methyl halides and between CH3I and both CHCl3 and C2HCl3 may indicate that the two processes occur simultaneously in the coastal area of the Dead Sea.
Although relatively high, the CHBr3 fluxes and mixing ratios that we measured at the Dead Sea cannot be directly related to the high mixing ratios of reactive bromine species that were found at the Dead Sea (e.g., Matveev et al., 2001; Tas et al., 2005) via its photolysis. Similarly, if CH3I photolysis is the only source of reactive I species, the measured fluxes and elevated mixing ratios of CH3I are not high enough to account for the high iodine monoxide in this area. Given their relatively fast photolysis, however, CH3I and CHBr3, as well as CH2Br2, may well have roles in the initiation of reactive bromine and iodine formation in this area.
Overall, along with other studies, the findings presented here highlight the potentially important role of saline soil and salt lakes in VHOC emission and call for further research on VHOC emission rates and controlling mechanisms and implications on stratospheric and tropospheric chemistry.
Data are available upon request from the corresponding author Eran Tas (eran.tas@mail.huji.ac.il).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-7667-2019-supplement.
ET, AG, RR and AW designed the experiments. MS, GL and QL carried out the field measurements, and DB carried out the sampled air analyses. GL contributed to designing and constructing a special mechanism for the simultaneous lifting and dropping of sampling canisters. Data curation and formal analysis were performed by ET and MS, with support from RR. ET and MS prepared the paper, with contributions from all co-authors.
The authors declare that they have no conflict of interest.
We thank the United States–Israel Binational Science Foundation (grant no. 2012287) for funding this study. Eran Tas holds the Joseph H. and Belle R. Braun Senior Lectureship in Agriculture.
This research has been supported by the United States–Israel Binational Science Foundation (grant no. 2012287).
This paper was edited by James Roberts and reviewed by two anonymous referees.
Albers, C. N., Jacobsen, O. S., Flores, E. M. M., and Johnsen, A. R.: Arctic and Subarctic Natural Soils Emit Chloroform and Brominated Analogues by Alkaline Hydrolysis of Trihaloacetyl Compounds, Environ. Sci. Technol., 51, 6131–6138, https://doi.org/10.1021/acs.est.7b00144, 2017.
Alpert, P., Shafir, H., and Issahary, D.: Recent changes in the climate at the dead sea – A preliminary study, Climatic Change, 37, 513–537, https://doi.org/10.1023/A:1005330908974, 1997.
Bondu, S., Cocquempot, B., Deslandes, E., and Morin, P.: Effects of salt and light stress on the release of volatile halogenated organic compounds by Solieria chordalis: a laboratory incubation study, Bot. Mar., 51, 485–492, https://doi.org/10.1515/Bot.2008.056, 2008.
Breider, F. and Albers, C. N.: Formation mechanisms of trichloromethyl-containing compounds in the terrestrial environment: A critical review, Chemosphere, 119, 145–154, https://doi.org/10.1016/j.chemosphere.2014.05.080, 2015.
Brinckmann, S., Engel, A., Bönisch, H., Quack, B., and Atlas, E.: Short-lived brominated hydrocarbons – observations in the source regions and the tropical tropopause layer, Atmos. Chem. Phys., 12, 1213–1228, https://doi.org/10.5194/acp-12-1213-2012, 2012.
BS 1377-3: Methods of Test for Soils for Civil Engineering Purposes – Part 3: Chemical and Electro-chemical Tests. British Standards Institution, London, UK, 1990.
Buchalo, A. S., Nevo, E., Wasser, S. P., Oren, A., and Molitoris, H. P.: Fungal life in the extremely hypersaline water of the Dead Sea: first records, P. Roy. Soc. B-Biol. Sci., 265, 1461–1465, https://doi.org/10.1098/rspb.1998.0458, 1998.
Butler, J. H., King, D. B., Lobert, J. M., Montzka, S. A., Yvon-Lewis, S. A., Hall, B. D., Warwick, N. J., Mondeel, D. J., Aydin, M., and Elkins, J. W.: Oceanic distributions and emissions of short-lived halocarbons, Global Biogeochem. Cy., 21, Gb1023, https://doi.org/10.1029/2006gb002732, 2007.
Carpenter, L. J., Green, T. J., Mills, G. P., Bauguitte, S., Penkett, S. A., Zanis, P., Schuepbach, E., Schmidbauer, N., Monks, P. S., and Zellweger, C.: Oxidized nitrogen and ozone production efficiencies in the springtime free troposphere over the Alps, J. Geophys. Res.-Atmos., 105, 14547–14559, https://doi.org/10.1029/2000jd900002, 2000.
Carpenter, L. J., Wevill, D. J., O'Doherty, S., Spain, G., and Simmonds, P. G.: Atmospheric bromoform at Mace Head, Ireland: seasonality and evidence for a peatland source, Atmos. Chem. Phys., 5, 2927–2934, https://doi.org/10.5194/acp-5-2927-2005, 2005.
Carpenter, L. J., Jones, C. E., Dunk, R. M., Hornsby, K. E., and Woeltjen, J.: Air-sea fluxes of biogenic bromine from the tropical and North Atlantic Ocean, Atmos. Chem. Phys., 9, 1805–1816, https://doi.org/10.5194/acp-9-1805-2009, 2009.
Carpenter, L. J., MacDonald, S. M., Shaw, M. D., Kumar, R., Saunders, R. W., Parthipan, R., Wilson, J., and Plane, J. M. C.: Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine, Nat. Geosci., 6, 108–111, https://doi.org/10.1038/Ngeo1687, 2013.
Carpenter, L. J., Reimann, S., Burkholder, J. B., Clerbaux, C., Hall, B. D., Hossaini, R., Laube, J. C., and Yvon-Lewis, S. A.: Ozone-depleting substances (ODSs) and other gases of interest to the Montreal Protocol, Chapter 1 in Scientific Assessment of Ozone Depletion: 2014, Global Ozone Research and Monitoring Project – Report No. 55, World Meteorological Organization, Geneva, Switzerland, 2014.
Colman, J. J., Swanson, A. L., Meinardi, S., Sive, B. C., Blake, D. R., and Rowland, F. S.: Description of the analysis of a wide range of volatile organic compounds in whole air samples collected during PEM-Tropics A and B, Anal. Chem., 73, 3723–3731, https://doi.org/10.1021/ac010027g, 2001.
Dearellano, J. V. G., Duynkerke, P. G., and Zeller, K. F.: Atmospheric Surface-Layer Similarity Theory Applied to Chemically Reactive Species, J. Geophys. Res.-Atmos., 100, 1397–1408, https://doi.org/10.1029/94jd02434, 1995.
Derendorp, L., Wishkerman, A., Keppler, F., McRoberts, C., Holzinger, R., and Rockmann, T.: Methyl chloride emissions from halophyte leaf litter: Dependence on temperature and chloride content, Chemosphere, 87, 483–489, https://doi.org/10.1016/j.chemosphere.2011.12.035, 2012.
Deventer, M. J., Jiao, Y., Knox, S. H., Anderson, F., Ferner, M. C., Lewis, J. A., and Rhew, R. C.: Ecosystem-Scale Measurements of Methyl Halide Fluxes From a Brackish Tidal Marsh Invaded With Perennial Pepperweed (Lepidium latifolium), J. Geophys. Res.-Biogeo., 123, 2104–2120, https://doi.org/10.1029/2018JG004536, 2018.
Dimmer, C. H., Simmonds, P. G., Nickless, G., and Bassford, M. R.: Biogenic fluxes of halomethanes from Irish peatland ecosystems, Atmos. Environ., 35, 321–330, https://doi.org/10.1016/S1352-2310(00)00151-5, 2001.
Dyer, A. J. and Bradley, E. F.: An Alternative Analysis of Flux-Gradient Relationships at the 1976 Itce, Bound.-Lay. Meteorol., 22, 3–19, https://doi.org/10.1007/Bf00128053, 1982.
Ekdahl, A., Pedersen, M., and Abrahamsson, K.: A study of the diurnal variation of biogenic volatile halocarbons, Mar. Chem., 63, 1–8, https://doi.org/10.1016/S0304-4203(98)00047-4, 1998.
Gan, J., Yates, S. R., Ohr, H. D., and Sims, J. J.: Production of methyl bromide by terrestrial higher plants, Geophys. Res. Lett., 25, 3595–3598, https://doi.org/10.1029/98gl52697, 1998.
Gebhardt, S., Colomb, A., Hofmann, R., Williams, J., and Lelieveld, J.: Halogenated organic species over the tropical South American rainforest, Atmos. Chem. Phys., 8, 3185–3197, https://doi.org/10.5194/acp-8-3185-2008, 2008.
Gifford, F. A.: Turbulent diffusion typing schemes: A review, under Consequences of Effluent Release, edited by: Schoup, R. L., Nucl. Safety, 17, 68–86, 1976.
Golder, D.: Relations among stability parameters in the surface layer, 3, 47–58, Bound.-Lay. Meteorol., 31, 47–58, 1972.
Grubbs, F. E. and Beck, G.: Extension of Sample Sizes and Percentage Points for Significance Tests of Outlying Observations, Technometrics, 14, 847–854, https://doi.org/10.2307/1267134, 1972.
Gualtieri, G. and Secci, S.: Comparing methods to calculate atmospheric stability-dependent wind speed profiles: A case study on coastal location, Renew. Energ., 36, 2189–2204, https://doi.org/10.1016/j.renene.2011.01.023, 2011.
Hebestreit, K., Stutz, J., Rosen, D., Matveiv, V., Peleg, M., Luria, M., and Platt, U.: DOAS measurements of tropospheric bromine oxide in mid-latitudes, Science, 283, 55–57, https://doi.org/10.1126/science.283.5398.55, 1999.
Hoekstra, E. J., De Leer, E. W. B., and Brinkman, U. A. T.: Natural formation of chloroform and brominated trihalomethanes in soil, Environ. Sci. Technol., 32, 3724–3729, https://doi.org/10.1021/es980127c, 1998.
Hossaini, R., Chipperfield, M. P., Monge-Sanz, B. M., Richards, N. A. D., Atlas, E., and Blake, D. R.: Bromoform and dibromomethane in the tropics: a 3-D model study of chemistry and transport, Atmos. Chem. Phys., 10, 719–735, https://doi.org/10.5194/acp-10-719-2010, 2010.
Hossaini, R., Mantle, H., Chipperfield, M. P., Montzka, S. A., Hamer, P., Ziska, F., Quack, B., Krüger, K., Tegtmeier, S., Atlas, E., Sala, S., Engel, A., Bönisch, H., Keber, T., Oram, D., Mills, G., Ordóñez, C., Saiz-Lopez, A., Warwick, N., Liang, Q., Feng, W., Moore, F., Miller, B. R., Marécal, V., Richards, N. A. D., Dorf, M., and Pfeilsticker, K.: Evaluating global emission inventories of biogenic bromocarbons, Atmos. Chem. Phys., 13, 11819–11838, https://doi.org/10.5194/acp-13-11819-2013, 2013.
Huber, S. G., Kotte, K., Scholer, H. F., and Williams, J.: Natural Abiotic Formation of Trihalomethanes in Soil: Results from Laboratory Studies and Field Samples, Environ. Sci. Technol., 43, 4934–4939, https://doi.org/10.1021/es8032605, 2009.
IPCC: Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Solomon, S., Qin, D., Manning, M., Chen, Z., Marquis, M., Averyt, K. B., Tignor, M., and Miller, H. L., Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2007.
Jacob, J. H., Hussein, E. I., Shakhatreh, M. A. K., and Cornelison, C. T.: Microbial community analysis of the hypersaline water of the Dead Sea using high-throughput amplicon sequencing, Microbiologyopen, 6, e500, https://doi.org/10.1002/mbo3.500, 2017.
Jarraud, M.: Guide to Meteorological Instruments and Methods of Observation., 7th ed., WMO8, 681 pp., 2008
Jiao, Y., Ruecker, A., Deventer, M. J., Chow, A. T., and Rhew, R. C.: Halocarbon emissions from a degraded forested wetland in coastal South Carolina impacted by sea level rise, ACS Earth Space Chem., 2, 955–967, https://doi.org/10.1021/acsearthspacechem.8b00044, 2018.
Keppler, F., Eiden, R., Niedan, V., Pracht, J., and Scholer, H. F.: Halocarbons produced by natural oxidation processes during degradation of organic matter, Nature, 403, 298–301, https://doi.org/10.1038/35002055, 2000.
Khan, M. A. H., Whelan, M. E., and Rhew, R. C.: Effects of temperature and soil moisture on methyl halide and chloroform fluxes from drained peatland pasture soils, J. Environ. Monitor., 14, 241–249, https://doi.org/10.1039/c1em10639b, 2012.
Kis-Papo, T., Grishkan, I., Oren, A., Wasser, S. P., and Nevo, E.: Spatiotemporal diversity of filamentous fungi in the hypersaline Dead Sea, Mycol. Res., 105, 749–756, https://doi.org/10.1017/S0953756201004129, 2001.
Kotte, K., Löw, F., Huber, S. G., Krause, T., Mulder, I., and Schöler, H. F.: Organohalogen emissions from saline environments – spatial extrapolation using remote sensing as most promising tool, Biogeosciences, 9, 1225–1235, https://doi.org/10.5194/bg-9-1225-2012, 2012.
Kuyper, B., Palmer, C. J., Labuschagne, C., and Reason, C. J. C.: Atmospheric bromoform at Cape Point, South Africa: an initial fixed-point data set on the African continent, Atmos. Chem. Phys., 18, 5785–5797, https://doi.org/10.5194/acp-18-5785-2018, 2018.
Lee-Taylor, J. M. and Holland, E. A.: Litter decomposition as a potential natural source of methyl bromide, J. Geophys. Res.-Atmos., 105, 8857–8864, https://doi.org/10.1029/1999jd901112, 2000.
Lenschow, D. H.: Micrometeorological techniques for measuring biosphere-atmosphere trace gas exchange, Biogenic Trace Gases: Measuring Emissions from Soil and Water, edited by: Matson, P. A. and Hariss, R. C., Methods in Ecology, Blackwell Science, Oxford, 126–163, 1995.
Liu, Y. N., Yvon-Lewis, S. A., Hu, L., Salisbury, J. E., and O'Hern, J. E.: CHBr3, CH2Br2, and CHClBr2 in U.S. coastal waters during the Gulf of Mexico and East Coast Carbon cruise, J. Geophys. Res.-Oceans, 116, C10004, https://doi.org/10.1029/2010jc006729, 2011.
Maier, M. and Schack-Kirchner, H.: Using the gradient method to determine soil gas flux: A review, Agr. Forest Meteorol., 192, 78–95, https://doi.org/10.1016/j.agrformet.2014.03.006, 2014.
Manley, S. L. and Dastoor, M. N.: Methyl-Iodide (Ch3i) Production by Kelp and Associated Microbes, Mar. Biol., 98, 477–482, https://doi.org/10.1007/Bf00391538, 1988.
Manley, S. L., Wang, N. Y., Walser, M. L., and Cicerone, R. J.: Coastal salt marshes as global methyl halide sources from determinations of intrinsic production by marsh plants, Global Biogeochem. Cy., 20, Gb3015, https://doi.org/10.1029/2005gb002578, 2006.
Matveev, V., Peleg, M., Rosen, D., Tov-Alper, D. S., Hebestreit, K., Stutz, J., Platt, U., Blake, D., and Luria, M.: Bromine oxide – ozone interaction over the Dead Sea, J. Geophys. Res.-Atmos., 106, 10375–10387, https://doi.org/10.1029/2000jd900611, 2001.
Meredith, L. K., Commane, R., Munger, J. W., Dunn, A., Tang, J., Wofsy, S. C., and Prinn, R. G.: Ecosystem fluxes of hydrogen: a comparison of flux-gradient methods, Atmos. Meas. Tech., 7, 2787–2805, https://doi.org/10.5194/amt-7-2787-2014, 2014.
Mohd Nadzir, M. S., Phang, S. M., Abas, M. R., Abdul Rahman, N., Abu Samah, A., Sturges, W. T., Oram, D. E., Mills, G. P., Leedham, E. C., Pyle, J. A., Harris, N. R. P., Robinson, A. D., Ashfold, M. J., Mead, M. I., Latif, M. T., Khan, M. F., Amiruddin, A. M., Banan, N., and Hanafiah, M. M.: Bromocarbons in the tropical coastal and open ocean atmosphere during the 2009 Prime Expedition Scientific Cruise (PESC-09), Atmos. Chem. Phys., 14, 8137–8148, https://doi.org/10.5194/acp-14-8137-2014, 2014.
Moore, R. M.: Methyl halide production and loss rates in sea water from field incubation experiments, Mar. Chem., 101, 213–219, https://doi.org/10.1016/j.marchem.2006.03.003, 2006.
Moore, R. M., Gut, A., and Andreae, M. O.: A pilot study of methyl chloride emissions from tropical woodrot fungi, Chemosphere, 58, 221–225, https://doi.org/10.1016/j.chemosphere.2004.03.011, 2005.
Niemi, T. M., Ben-Avrahem, Z., and Gat, J. R.: The Dead Sea: The Lake and Its Setting, Oxford Monogr. Geol. Geophys., vol. 36, Oxford Univ. Press, New York, 1997.
O'Brien, L. M., Harris, N. R. P., Robinson, A. D., Gostlow, B., Warwick, N., Yang, X., and Pyle, J. A.: Bromocarbons in the tropical marine boundary layer at the Cape Verde Observatory – measurements and modelling, Atmos. Chem. Phys., 9, 9083–9099, https://doi.org/10.5194/acp-9-9083-2009, 2009.
Obrist, D., Tas, E., Peleg, M., Matveev, V., Fain, X., Asaf, D., and Luria, M.: Bromine-induced oxidation of mercury in the mid-latitude atmosphere, Nat. Geosci., 4, 22–26, https://doi.org/10.1038/Ngeo1018, 2011.
O'Dowd, C. D., Jimenez, J. L., Bahreini, R., Flagan, R. C., Seinfeld, J. H., Hameri, K., Pirjola, L., Kulmala, M., Jennings, S. G., and Hoffmann, T.: Marine aerosol formation from biogenic iodine emissions, Nature, 417, 632–636, https://doi.org/10.1038/nature00775, 2002.
Oren, A. and Shilo, M.: Factors Determining the Development of Algal and Bacterial Blooms in the Dead-Sea – a Study of Simulation Experiments in Outdoor Ponds, FEMS Microbiol. Ecol., 31, 229–237, 1985.
Oren, A., Ionescu, D., Hindiyeh, M., and Malkawi, H.: Microalgae and cyanobacteria of the Dead Sea and its surrounding springs, Isr. J. Plant Sci., 56, 1–13, https://doi.org/10.1560/Ijps.56.1-2.1, 2008.
Osman, K. T.: Forest soils: properties and management, in: Physical properties of forest soils, Springer International Publishing, 19–28, 2013.
Pasquill, F. and Smith, F. B.: The physical and meteorological basis for the estimation of the dispersion of windborn material, in: Proceedings of the Second International Clean Air Congress, edited by: Englund, H. M. and Beery, W. T., Washington, DC, 1970, 1067–1072, Academic Press, New York, 1971.
Pedersen, M., Collen, J., Abrahamsson, K., and Ekdahl, A.: Production of halocarbons from seaweeds: An oxidative stress reaction?, Sci. Mar., 60, 257–263, 1996.
Pen-Mouratov, S., Myblat, T., Shamir, I., Barness, G., and Steinberger, Y.: Soil Biota in the Arava Valley of Negev Desert, Israel, Pedosphere, 20, 273–284, https://doi.org/10.1016/S1002-0160(10)60015-X, 2010.
Pyle, J. A., Ashfold, M. J., Harris, N. R. P., Robinson, A. D., Warwick, N. J., Carver, G. D., Gostlow, B., O'Brien, L. M., Manning, A. J., Phang, S. M., Yong, S. E., Leong, K. P., Ung, E. H., and Ong, S.: Bromoform in the tropical boundary layer of the Maritime Continent during OP3, Atmos. Chem. Phys., 11, 529–542, https://doi.org/10.5194/acp-11-529-2011, 2011.
Quack, B. and Wallace, D. W. R.: Air-sea flux of bromoform: Controls, rates, and implications (vol 17, art no 1023, 2003), Global Biogeochem. Cy., 18, Gb1004, https://doi.org/10.1029/2003gb002187, 2004.
Quack, B., Atlas, E., Petrick, G., and Wallace, D. W. R.: Bromoform and dibromomethane above the Mauritanian upwelling: Atmospheric distributions and oceanic emissions, J. Geophys. Res.-Atmos., 112, D09312, https://doi.org/10.1029/2006jd007614, 2007.
Rhew, R. C., Miller, B. R., and Weiss, R. F.: Natural methyl bromide and methyl chloride emissions from coastal salt marshes, Nature, 403, 292–295, https://doi.org/10.1038/35002043, 2000.
Rhew, R. C., Miller, B. R., Vollmer, M. K., and Weiss, R. F.: Shrubland fluxes of methyl bromide and methyl chloride, J. Geophys. Res.-Atmos., 106, 20875–20882, https://doi.org/10.1029/2001jd000413, 2001.
Rhew, R. C., Miller, B. R., Bill, M., Goldstein, A. H., and Weiss, R. F.: Environmental and biological controls on methyl halide emissions from southern California coastal salt marshes, Biogeochemistry, 60, 141–161, https://doi.org/10.1023/A:1019812006560, 2002.
Rhew, R. C., Aydin, M., and Saltzman, E. S.: Measuring terrestrial fluxes of methyl chloride and methyl bromide using a stable isotope tracer technique, Geophys. Res. Lett., 30, 2103, https://doi.org/10.1029/2003gl018160, 2003.
Rhew, R. C., Teh, Y. A., Abel, T., Atwood, A., and Mazeas, O.: Chloroform emissions from the Alaskan Arctic tundra, Geophys. Res. Lett., 35, L21811, https://doi.org/10.1029/2008gl035762, 2008.
Rhew, R. C., Whelan, M. E., and Min, D.-H.: Large methyl halide emissions from south Texas salt marshes, Biogeosciences, 11, 6427–6434, https://doi.org/10.5194/bg-11-6427-2014, 2014.
Rousseaux, M. C., Ballare, C. L., Giordano, C. V., Scopel, A. L., Zima, A. M., Szwarcberg-Bracchitta, M., Searles, P. S., Caldwell, M. M., and Diaz, S. B.: Ozone depletion and UVB radiation: Impact on plant DNA damage in southern South America, P. Natl. Acad. Sci. USA, 96, 15310–15315, https://doi.org/10.1073/pnas.96.26.15310, 1999.
Ruecker, A., Weigold, P., Behrens, S., Jochmann, M., Laaks, J., and Kappler, A.: Predominance of Biotic over Abiotic Formation of Halogenated Hydrocarbons in Hypersaline Sediments in Western Australia, Environ. Sci. Technol., 48, 9170–9178, https://doi.org/10.1021/es501810g, 2014.
Schmugge, T. J. and André, J.-C.: Land surface evaporation: measurement and parameterization: Springer Science & Business Media, 1991.
Shafir, H. and Alpert, P.: Regional and local climatic effects on the Dead-Sea evaporation, Clim. Change, 105, 455–468, https://doi.org/10.1007/s10584-010-9892-8, 2010.
Simmonds, P. G., Derwent, R. G., Manning, A. J., O'Doherty, S., and Spain, G.: Natural chloroform emissions from the blanket peat bogs in the vicinity of Mace Head, Ireland over a 14-year period, Atmos. Environ., 44, 1284–1291, https://doi.org/10.1016/j.atmosenv.2009.12.027, 2010.
Simpson, W. R., Brown, S. S., Saiz-Lopez, A., Thornton, J. A., and von Glasow, R.: Tropospheric Halogen Chemistry: Sources, Cycling, and Impacts, Chem. Rev., 115, 4035–4062, https://doi.org/10.1021/cr5006638, 2015.
Sive, B. C., Varner, R. K., Mao, H., Blake, D. R., Wingenter, O. W., and Talbot, R.: A large terrestrial source of methyl iodide, Geophys. Res. Lett., 34, L17808, https://doi.org/10.1029/2007gl030528, 2007.
Stull, R. B.: An introduction to boundary layer meteorology, Kluwer, Dordrecht, 1988.
Sverdrup, H. U., Johnson, M. W., and Fleming, R. H.: The Oceans, Their Physics, Chemistry and General Biology, Prentice-Hall, Englewood Cliffs, N.J., 1942.
Tas, E., Matveev, V., Zingler, J., Luria, M., and Peleg, M.: Frequency and extent of ozone destruction episodes over the Dead Sea, Israel, Atmos. Environ., 37, 4769–4780, https://doi.org/10.1016/j.atmosenv.2003.08.015, 2003.
Tas, E., Peleg, M., Matveev, V., Zingler, J., and Luria, M.: Frequency and extent of bromine oxide formation over the Dead Sea, J. Geophys. Res.-Atmos., 110, D11304, https://doi.org/10.1029/2004jd005665, 2005.
Tas, E., Peleg, M., Pedersen, D. U., Matveev, V., Pour Biazar, A., and Luria, M.: Measurement-based modeling of bromine chemistry in the boundary layer: 1. Bromine chemistry at the Dead Sea, Atmos. Chem. Phys., 6, 5589–5604, https://doi.org/10.5194/acp-6-5589-2006, 2006.
Tas, E., Obrist, D., Peleg, M., Matveev, V., Faïn, X., Asaf, D., and Luria, M.: Measurement-based modelling of bromine-induced oxidation of mercury above the Dead Sea, Atmos. Chem. Phys., 12, 2429–2440, https://doi.org/10.5194/acp-12-2429-2012, 2012.
Teh, Y. A., Rhew, R. C., Atwood, A., and Abel, T.: Water, temperature, and vegetation regulation of methyl chloride and methyl bromide fluxes from a shortgrass steppe ecoystem, Glob. Change Biol., 14, 77–91, https://doi.org/10.1111/j.1365-2486.2007.01480.x, 2008.
Varner, R. K., Crill, P. M., and Talbot, R. W.: Wetlands: a potentially significant source of atmospheric methyl bromide and methyl chloride, Geophys. Res. Lett., 26, 2433–2435, https://doi.org/10.1029/1999gl900587, 1999.
Warwick, N. J., Pyle, J. A., and Shallcross, D. E.: Global modelling of the atmospheric methyl bromide budget, J. Atmos. Chem., 54, 133–159, https://doi.org/10.1007/s10874-006-9020-3, 2006.
Watling, R. and Harper, D. B.: Chloromethane production by wood-rotting fungi and an estimate of the global flux to the atmosphere, Mycol. Res., 102, 769–787, https://doi.org/10.1017/S0953756298006157, 1998.
Weissflog, L., Lange, C. A., Pfennigsdorff, A., Kotte, K., Elansky, N., Lisitzyna, L., Putz, E., and Krueger, G.: Sediments of salt lakes as a new source of volatile highly chlorinated C1/C2 hydrocarbons, Geophys. Res. Lett., 32, L01401, https://doi.org/10.1029/2004gl020807, 2005.
Wishkerman, A., Gebhardt, S., McRoberts, C. W., Hamilton, J. T. G., Williams, J., and Keppler, F.: Abiotic methyl bromide formation from vegetation, and its strong dependence on temperature, Environ. Sci. Technol., 42, 6837–6842, https://doi.org/10.1021/es800411j, 2008.
Xiao, X., Prinn, R. G., Fraser, P. J., Simmonds, P. G., Weiss, R. F., O'Doherty, S., Miller, B. R., Salameh, P. K., Harth, C. M., Krummel, P. B., Porter, L. W., Mühle, J., Greally, B. R., Cunnold, D., Wang, R., Montzka, S. A., Elkins, J. W., Dutton, G. S., Thompson, T. M., Butler, J. H., Hall, B. D., Reimann, S., Vollmer, M. K., Stordal, F., Lunder, C., Maione, M., Arduini, J., and Yokouchi, Y.: Optimal estimation of the surface fluxes of methyl chloride using a 3-D global chemical transport model, Atmos. Chem. Phys., 10, 5515–5533, https://doi.org/10.5194/acp-10-5515-2010, 2010.
Yang, K., Tamai, N., and Koike, T.: Analytical solution of surface layer similarity equations, J. Appl. Meteorol., 40, 1647–1653, https://doi.org/10.1175/1520-0450(2001)040<1647:Asosls>2.0.Co;2, 2001.
Yassaa, N., Wishkerman, A., Keppler, F., and Williams, J.: Fast determination of methyl chloride and methyl bromide emissions from dried plant matter and soil samples using HS-SPME and GC-MS: method and first results, Environ. Chem., 6, 311–318, https://doi.org/10.1071/En09034, 2009.
Yokouchi, Y., Ikeda, M., Inuzuka, Y., and Yukawa, T.: Strong emission of methyl chloride from tropical plants, Nature, 416, 163–165, https://doi.org/10.1038/416163a, 2002.
Yokouchi, Y., Inagaki, T., Yazawa, K., Tamaru, T., Enomoto, T., and Izumi, K.: Estimates of ratios of anthropogenic halocarbon emissions from Japan based on aircraft monitoring over Sagami Bay, Japan, J. Geophys. Res.-Atmos., 110, D06301, https://doi.org/10.1029/2004jd005320, 2005.
Yokouchi, Y., Saito, T., Ishigaki, C., and Aramoto, M.: Identification of methyl chloride-emitting plants and atmospheric measurements on a subtropical island, Chemosphere, 69, 549–553, https://doi.org/10.1016/j.chemosphere.2007.03.028, 2007.
Zhou, Y., Varner, R. K., Russo, R. S., Wingenter, O. W., Haase, K. B., Talbot, R., and Sive, B. C.: Coastal water source of short-lived halocarbons in New England, J. Geophys. Res.-Atmos., 110, D21302, https://doi.org/10.1029/2004jd005603, 2005.
Zhou, Y., Mao, H. T., Russo, R. S., Blake, D. R., Wingenter, O. W., Haase, K. B., Ambrose, J., Varner, R. K., Talbot, R., and Sive, B. C.: Bromoform and dibromomethane measurements in the seacoast region of New Hampshire, 2002–2004, J. Geophys. Res.-Atmos., 113, D08305, https://doi.org/10.1029/2007jd009103, 2008.
Zingler, J. and Platt, U.: Iodine oxide in the Dead Sea Valley: Evidence for inorganic sources of boundary layer IO, J. Geophys. Res.-Atmos., 110, D07307, https://doi.org/10.1029/2004jd004993, 2005.
Ziska, F., Quack, B., Abrahamsson, K., Archer, S. D., Atlas, E., Bell, T., Butler, J. H., Carpenter, L. J., Jones, C. E., Harris, N. R. P., Hepach, H., Heumann, K. G., Hughes, C., Kuss, J., Krüger, K., Liss, P., Moore, R. M., Orlikowska, A., Raimund, S., Reeves, C. E., Reifenhäuser, W., Robinson, A. D., Schall, C., Tanhua, T., Tegtmeier, S., Turner, S., Wang, L., Wallace, D., Williams, J., Yamamoto, H., Yvon-Lewis, S., and Yokouchi, Y.: Global sea-to-air flux climatology for bromoform, dibromomethane and methyl iodide, Atmos. Chem. Phys., 13, 8915–8934, https://doi.org/10.5194/acp-13-8915-2013, 2013.