the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 29 Nov 2019
| 29 Nov 2019
Long-term total OH reactivity measurements in a boreal forest
Arnaud P. Praplan
Toni Tykkä
Dean Chen
Michael Boy
Ditte Taipale
Ville Vakkari
Putian Zhou
Tuukka Petäjä
Heidi Hellén
Total hydroxyl radical (OH) reactivity measurements were conducted at the second Station for Measuring Ecosystem–Atmosphere Relations (SMEAR II), a boreal forest site located in Hyytiälä, Finland, from April to July 2016. The measured values were compared with OH reactivity calculated from a combination of data from the routine trace gas measurements (station mast) as well as online and offline analysis with a gas chromatographer coupled to a mass spectrometer (GC–MS) and offline liquid chromatography. Up to 104 compounds, mostly volatile organic compounds (VOCs) and oxidized VOCs, but also inorganic compounds, were included in the analysis, even though the data availability for each compound varied with time. The monthly averaged experimental total OH reactivity was found to be higher in April and May (ca. 20 s−1) than in June and July (7.6 and 15.4 s−1, respectively). The measured values varied much more in spring with high reactivity peaks in late afternoon, with values higher than in the summer, in particular when the soil was thawing. Total OH reactivity values generally followed the pattern of mixing ratios due to change of the boundary layer height. The missing reactivity fraction (defined as the difference between measured and calculated OH reactivity) was found to be high. Several reasons that can explain the missing reactivity are discussed in detail such as (1) missing measurements due to technical issues, (2) not measuring oxidation compounds of detected biogenic VOCs, and (3) missing important reactive compounds or classes of compounds with the available measurements. In order to test the second hypothesis, a one-dimensional chemical transport model (SOSAA) has been used to estimate the amount of unmeasured oxidation products and their expected contribution to the reactivity for three different short periods in April, May, and July. However, only a small fraction (<4.5 %) of the missing reactivity can be explained by modelled secondary compounds (mostly oxidized VOCs). These findings indicate that compounds measured but not included in the model as well as unmeasured primary emissions contribute the missing reactivity. In the future, non-hydrocarbon compounds from sources other than vegetation (e.g. soil) should be included in OH reactivity studies.
Please read the corrigendum first before continuing.
-
Notice on corrigendum
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
-
Article
(16359 KB)
-
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
- Article
(16359 KB) - Corrigendum
- BibTeX
- EndNote





Terrestrial vegetation is responsible for about 90 % of the emissions of biogenic volatile organic compounds (BVOCs) into the atmosphere (Guenther et al., 1995). Isoprene and monoterpenes are the most abundant BVOCs globally with 44 % and 17 % of total biogenic emissions, respectively (Guenther et al., 2012). These compounds are very reactive and their lifetimes range from minutes to hours, thus influencing tropospheric chemistry.
Total hydroxyl radical (OH) reactivity measurements can be used as a method to assess our understanding of tropospheric chemistry (Kovacs and Brune, 2001; Williams and Brune, 2015). Many observations of total OH reactivity have been performed in the past few decades and compared to calculated OH reactivity derived from known chemical composition of the atmosphere. While for urban environments the unexplained (or missing) reactivity fraction remains low, it is often more than 50 % in forested environments (see the review by Yang et al., 2016). Based on these observations, Ferracci et al. (2018) modelled the global OH reactivity, as well as hypothetical missing chemical sink, which was found to be mostly localized above forested areas and in a few areas with large anthropogenic emissions.
Large fractions of missing reactivity were first observed in a forest in northern Michigan (Di Carlo et al., 2004) and later also observed in other forested environments (e.g. Hansen et al., 2014; Nakashima et al., 2014; Ramasamy et al., 2016; Zannoni et al., 2016). Also, in the tropical forest of Borneo up to 70 % of the measured total OH reactivity remained unexplained (Edwards et al., 2013). In addition, Nölscher et al. (2016) identified a large difference of missing OH reactivity between the dry and wet seasons in the Amazon rainforest, with 79 % on average and between 5 % and 15 %, respectively. They then identified the forest floor as an important but poorly characterized source of OH reactivity, and Bourtsoukidis et al. (2018) recently identified strong sesquiterpene emissions from soil microorganisms at the same site.
Also in the boreal forest, which represents approximately one-third of the Earth's forested surface (Keenan et al., 2015), a large discrepancy was observed between the total measured OH reactivity and the reactivity calculated from individual compounds present in the forest air (Sinha et al., 2010; Nölscher et al., 2012). Up to 89 % of the measured total OH reactivity could not be explained for periods in which the forest experienced stress conditions (elevated temperature).
The two main assumptions for the missing reactivity are (1) missing primary emissions and (2) missing oxidation products from the emissions. Several studies have been conducted to investigate these hypotheses. Nölscher et al. (2013), for instance, found an increasing missing fraction of Norway spruce (Picea abies) emissions from about 15 % to 27 % in spring and early summer and up to 70 %–84 % in late summer and autumn. In contrast, Kim et al. (2011) found no significant unknown primary BVOC contributing to OH reactivity (for red oak, white pine, beech, and red maple) during their study period in July 2009 in a forest in Michigan. They also found that the missing reactivity from ambient measurement at this site could be explained by oxidation products from isoprene. Kaiser et al. (2016) found in an isoprene-dominated forest in Alabama that emissions and their modelled oxidation products reduced the unexplained reactivity to 5 %–20 % during the day and 20 %–32 % at night, and they attribute the missing reactivity to unmeasured primary emissions. Mao et al. (2012) also demonstrated that including modelled oxidation products in OH reactivity calculations reduces the difference with measurements significantly.
Sinha et al. (2010) and Nölscher et al. (2012) conducted their studies at the second Station for Measuring Ecosystem–Atmosphere Relations (SMEAR II; Hari and Kulmala, 2005) in Hyytiälä, Finland, for about 3 weeks in August 2008 and for about 3.5 weeks in July–August 2010, respectively, with the comparative reactivity method (CRM, Sinha et al., 2008). Mogensen et al. (2011) modelled the full year of OH reactivity at SMEAR II for 2008, based on modelled emissions, known chemistry, and environmental conditions. A comparison with results from Sinha et al. (2010) showed that compounds other than monoterpenes, isoprene, and methane contribute to only about 8 % of the measured OH reactivity. Taking all compounds into account, about 61 % of the OH reactivity remained unexplained on average during that period. Mogensen et al. (2015) also compared modelled reactivity at SMEAR II with OH reactivity measurements from Nölscher et al. (2012), using measured trace gases as input, but found on average about 65 % of unexplained reactivity, similarly to the previous study.
In order to investigate OH reactivity at SMEAR II in more detail, in particular its missing fraction and the seasonal variations which are often neglected for summer intensive campaigns, a new implementation of the CRM was developed at the Finnish Meteorological Institute (Praplan et al., 2017). It was installed at SMEAR II along with instrumentation to measure VOCs in spring and summer 2016.
2.1 Measurement site
Measurements were conducted at the boreal forest site SMEAR II (Hari and Kulmala, 2005; Ilvesniemi et al., 2009) in Hyytiälä, Finland (61∘51′ N, 24∘17′ E, 181 m a.s.l.). The site is located in a ca. 60-year-old managed conifer forest with modest height variation in the terrain. The stand is dominated by Scots pine (Pinus sylvestris L.) homogeneously for about 200 m in all directions, extending to the north for about 1.2 km. Tampere is the largest city near the station about 60 km to the S–SW.
The instruments were located inside a container in an opening about 115 m from the site mast, from which meteorological data as well as ozone (O3), nitrogen oxides (NOx), methane (CH4), carbon monoxide (CO) and sulfur dioxide (SO2) concentrations were retrieved to be included in the analysis. Proton-transfer-reaction mass spectrometer (PTR-MS) measurements of VOCs usually operated at the station mast were not operational during the measurement period and could not be used in this study.
In situ measurements of the total OH reactivity (Sect. 2.5) and of VOC concentrations (Sect. 2.2) were performed at the container, sampling outside air at a height of about 1.5 m (Fig. 1). Station data (from the mast, measurement towers, and soil) are open data under the Creative Commons 4.0 Attribution licence (CC BY 4.0) and were retrieved from the online SmartSMEAR interface (https://avaa.tdata.fi/web/smart/smear, last access: 30 July 2019, Junninen et al., 2009).
Temperature and relative humidity (RH) are taken at 4.2 m above ground on the mast, soil properties are an average of five locations throughout the site, and radiation and precipitation data are collected at 18 m height on a nearby tower.

Figure 1Orthophotograph of the SMEAR II station in Hyytiälä and its surroundings with the marked location of the station mast and the container where the measurements were performed. (Source: Land Survey of Finland Topographic Database 09/2018).
2.2 In situ measurements of volatile organic compounds
VOCs were measured with two in situ gas chromatographer–mass spectrometers (GC–MSs). The first GC–MS was used for the measurements of mono- and sesquiterpenes, isoprene, 2-methyl-3-butenol (MBO), and C5−10 aldehydes. With this GC–MS air was drawn at the flow rate of 1 L min−1 through a metre-long fluorinated ethylene propylene (FEP) inlet (i.d. 1∕8 in.) and for O3 removal (Hellén et al., 2012) through a metre-long heated (120 ∘C) stainless-steel tube (o.d. 1∕8 in.). VOCs were collected from a 40 mL min−1 subsample flow in the cold trap (Carbopack B/Tenax TA) of the thermal desorption unit (TurboMatrix, 650, PerkinElmer) connected to a gas chromatograph (Clarus 680, PerkinElmer) coupled to a mass spectrometer (Clarus SQ 8 T, PerkinElmer). A HP-5 column (60 m, i.d. 0.25 mm, film thickness 1 µm) was used for separation. The second GC–MS was used for the measurements of C4−8 alcohols and C2−7 volatile organic acids (VOAs). Samples were taken every other hour. The sampling time was 60 min. Samples were analysed in situ with a thermal desorption unit (Unity 2 + Air Server 2, Markes International Ltd, Llantrisant, UK) connected to a gas chromatograph (Agilent 7890A, Agilent Technologies, Santa Clara, CA, USA) and a mass spectrometer (Agilent 5975C, Agilent Technologies, Santa Clara, CA, USA). A polyethylene glycol column DB-WAXetr (30 m, i.d. 0.25 mm, a film thickness 0.25 µm) was used for the separation. These methods and measurements have been described in more detail by Hellén et al. (2017, 2018).
2.3 Offline measurements of volatile organic compounds
Additional sampling took place between 27 April and 3 May in canisters and through adsorption cartridges (24 h time resolution) to be analysed by a GC–flame ionization detector (FID) (C2−6 hydrocarbons) and liquid chromatography with an ultraviolet absorption detector (LC–UV) (carbonyls), respectively. During this period, Tenax tube samples were also taken (4 h time resolution) and analysed later in the laboratory with GC–MS. These results were used as backup to fill in data during interruptions of the online GC–MS measurements. Between 20 and 29 July, additional sampling through adsorption cartridges for offline analysis with LC–UV was performed.
2.4 Mixing layer height measurements
The mixing layer height (MLH) was estimated from measurements with a 1.5 µm pulsed Doppler lidar (Halo Photonics Stream Line; Pearson et al., 2009) similar to Hellén et al. (2018). MLH was determined from a combination of turbulent kinetic energy dissipation rate profiles and conical scanning at a 30 ∘ elevation angle according to the method described in Vakkari et al. (2015). With this method MLH could be determined from 60 m a.g.l. (above ground level) to more than 2000 m a.g.l. at SMEAR II. Periods when MLH was <60 m a.g.l. could be identified, although the actual MLH was not determined due to minimum range limitations. MLH was not determined for rainy periods. For more detailed specifications of the lidar system and the applied MLH determination method; see Hellén et al. (2018).
2.5 Total OH reactivity measurements: the comparative reactivity method (CRM)
The OH reactivity, ROH, is defined as the sum of the concentration of individual compounds Xi multiplied by their respective reaction rate coefficient with respect to OH (). This can be summarized by the following equation:
The OH reactivity of a compound is the inverse of the OH chemical lifetime due to its reaction with that compound. High OH reactivity values correspond to short lifetimes, and long-lived species (such as methane) have a low reactivity.
Our analysis includes up to over 100 individual species from two GC–MS, GC–FID and LC–UV measurements (see Sect. 2.2 and 2.3). However, not all compounds have been measured at all times (see Fig. 6c). In addition NOx, O3, SO2, and CO concentrations were retrieved from the mast of the SMEAR II station, about 115 m away from the sampling position of total OH reactivity and VOCs.
Measurements of total OH reactivity (Rexp) have been conducted using the comparative reactivity method (CRM; Sinha et al., 2008; Michoud et al., 2015). Our particular implementation of the method is described in Praplan et al. (2017).
The CRM is based on the monitoring of pyrrole (C4H5N) mixed in a 100 mL reactor with zero air and ambient air, alternatively. The total flow through the reactor is about 465 mL min−1 and the residence time in the reactor is estimated at about 12–15 s.
Pyrrole detection is performed with a gas chromatograph (GC) equipped with a photon ionization detector (PID) every 2 min (Synthec Spectras GC955, Synspec BV, Groningen, the Netherlands). The sensitivity of this detector is independent of the RH of the sample (Fig. 2), but decreased from 1797 ppbv−1 (data from April to June) to 1290 ppbv−1 (July data). In both cases, the uncertainty of the sensitivity (Us) is 2.5 %. In addition we consider the uncertainty of the pyrrole levels (Up) based on the uncertainty of the pyrrole standard (Upyr,std = 10 %) and of the uncertainty of the dilution (Udil=8 %), so % (Praplan et al., 2017).
Based on earlier tests, the sensitivity of the GC–PID does not depend on RH in the reactor (Fig. 2, right). The sensitivity differs by only 2.8 % between humid and dry conditions, which is roughly the uncertainty of the sensitivity.
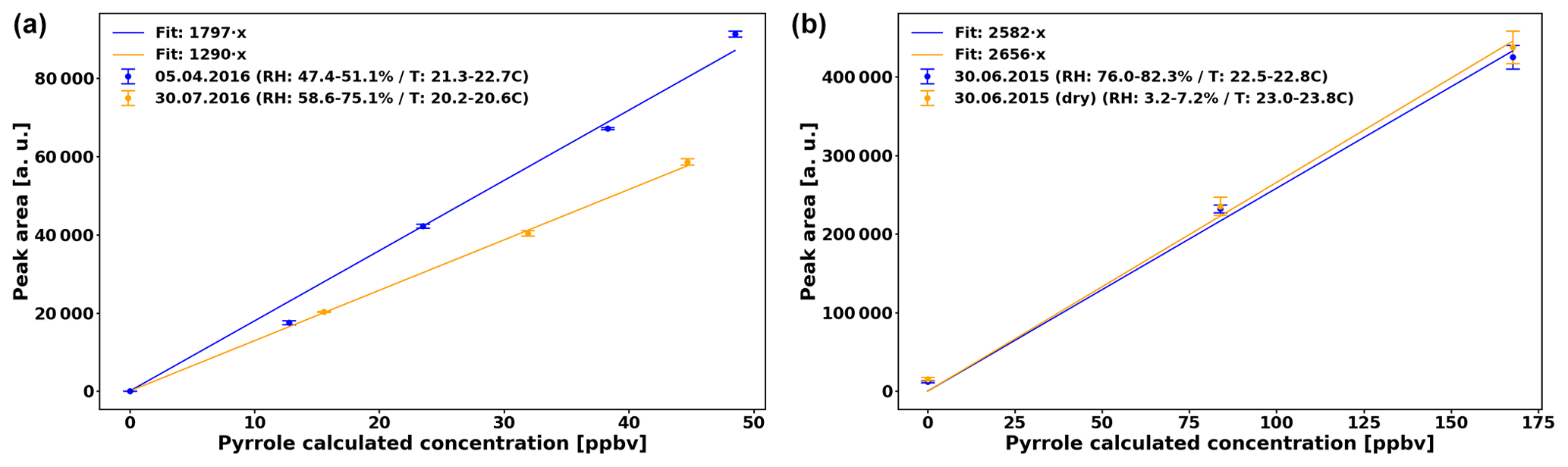
Figure 2(a) Sensitivity of the GC–PID for pyrrole used in the present study. (b) Same day (30 June 2015) sensitivity test for sensitivity of GC–PID for pyrrole in humid and dry conditions.
OH is produced by the photolysis of water (H2O) in a nitrogen flow (99.9999 % N2) using ultraviolet (UV) radiation and introduced into the CRM instrument reactor. Note that hydroperoxyl radicals (HO2) are concurrently produced from the reaction of hydrogen (H) with molecular oxygen (O2). In the zero air mixture, all OH radicals are consumed by pyrrole (C2 level), while ambient air contains other reactive compounds that compete for OH, leading to a higher pyrrole concentration (C3 level). The instrument switches between measurement of zero air and ambient air every 8 min. Stabilization of the conditions takes a couple of minutes and the first data point after each switch is discarded. From the difference between C2 and C3 pyrrole levels and taking into account the amount of pyrrole in the reactor in the absence of OH (C1, typically between 26 and 43 ppbv), the total OH reactivity Reqn can be derived from the following equation:
with kp the reaction rate of pyrrole with OH ( cm3 s−1; Atkinson et al., 1985). C1 is measured by introducing a large concentration of 0.6 % propane (C3H8) in nitrogen (N2) to act as an OH scavenger (Zannoni et al., 2015). Therefore, C1 takes into account the photolysis of pyrrole due to the UV radiation entering the reactor (8 %–13 %), which decreases the pyrrole concentration from the total amount of pyrrole injected in the reactor (C0 level).
Equation 2 assumes that OH levels are identical during C2 and C3 measurements. Therefore, variations in RH within the reactor but also the presence of O3 need to be taken into account. Therefore C3 in Eq. 2 results from the following:
with C3,exp the measured level of pyrrole in C3 mode, the correction due to different RH in C2 and C3 (the difference in RH is 4 % or less 99 % of the time, which corresponds to a change of no more than 5 % for Reqn), and the correction due to the presence in the reactor of additional O3 from sampled air. This last correction is discussed in detail in Sect. 2.5.1.
In addition, because of the dilution of the sampled air with humid nitrogen, the experimental total OH reactivity (Rexp) is derived from the following equation:
with D the dilution factor (ratio of sampling flow over total flow through the reactor) and Rmeasured the reactivity inside the reactor after applying corrections to Reqn (see Sect. 2.5.2).
Finally, the missing fraction of the total OH reactivity is obtained by comparing Rexp with ROH:
2.5.1 Ozone correction factor
As discussed in Praplan et al. (2017) and by Fuchs et al. (2017) for the CRM system of the Max Planck Institute, the pyrrole signal obtained during analysis of ambient air must be corrected for the presence of O3. Even though O3 is present in the reactor due to the UV lamp used to produce OH (about 170 ppbv in the instrument used in the present study), additional O3 sampled from ambient air (up to a 30 % increase) affects the OH concentration in the reactor, most probably by being photolysed and producing O(1D), which reacts further with H2O, yielding two OH radicals.
Praplan et al. (2017) used a correction (ΔC) independent of pyr ∕ OH as the experimental pyr ∕ OH for the measurements was in a narrow range close to 2. However as pyr ∕ OH varied from 1.0 to 5.3 in this study, a pyr ∕ OH-dependent correction has been derived.
The corrections ΔC were derived experimentally for various pyr ∕ OH values by injecting a known amount of O3 in the CRM's reactor (Fig. 3a) and then the slope of the linear fit (through the origin) for each pyr ∕ OH () was plotted against pyr ∕ OH (Fig. 3b). Based on these data, a linear fit has been derived to calculate according to pyr ∕ OH, and the uncertainty of this correction () is 30.8 %. When correcting ambient data in this study, the correction for a pyr ∕ OH of 3 ( = 0.079) has been applied when pyr ∕ OH was higher than 3 due to the lack of experimental data at higher pyr ∕ OH values. Note that a couple of experiments (with pyr ∕ OH 1.27 and 1.05) were performed with additional injection of propane (C3H8) as the pyrrole signal would have decreased to zero otherwise and no ΔC could have been determined.
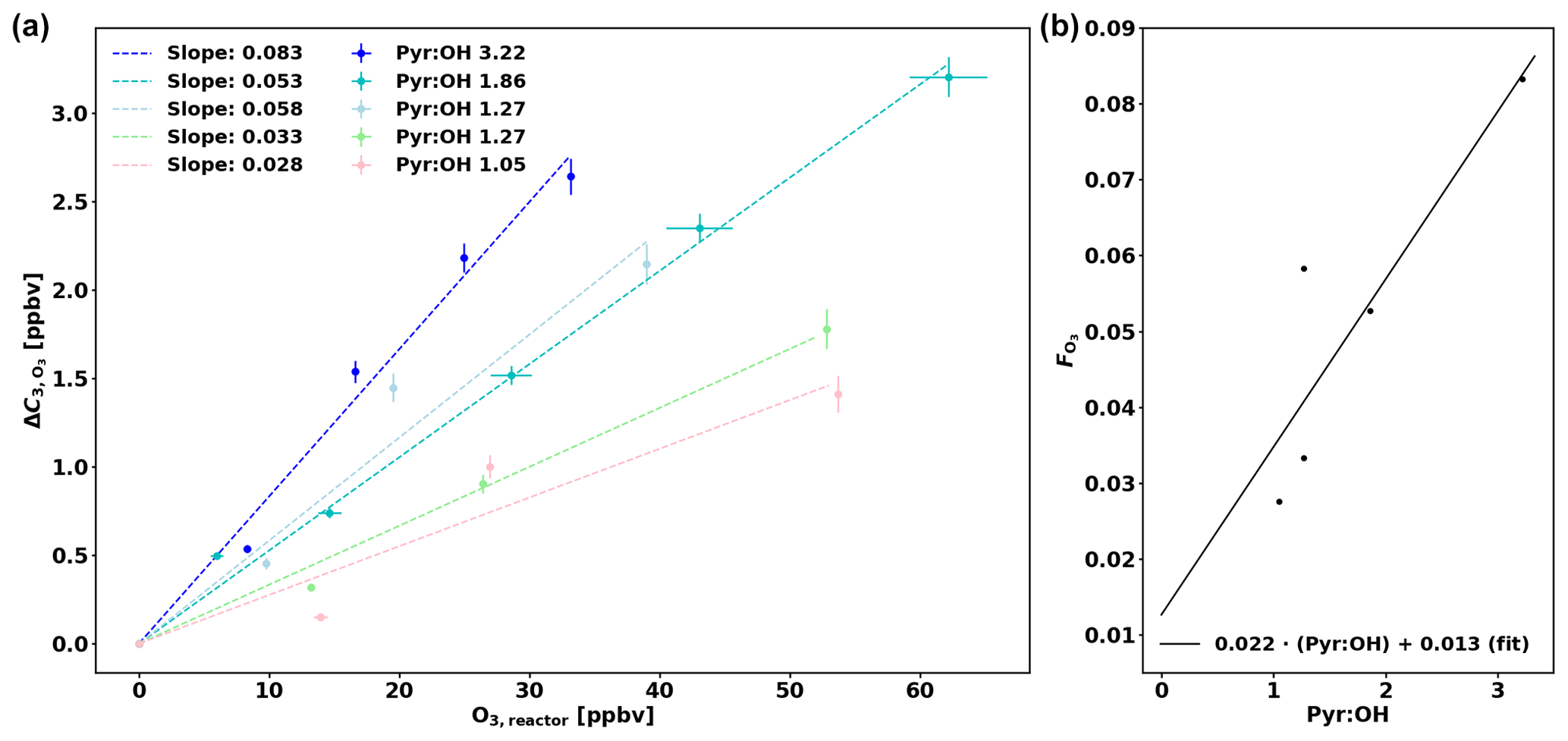
Figure 3(a) Correction of C3 (ΔC) as a function of ozone in the reactor (O3,reactor). (b) O3 correction factor () as a function of pyr ∕ OH.
The correction ΔC is then derived from the following equation:
As observed in Praplan et al. (2017), inhomogeneity of the air composition at the sampling site can affect the comparison between experimental total OH reactivity and calculated reactivity from known composition. It can for instance be directly affected by meteorology or changes in concentrations between the various sampling locations due to local emissions during low mixing periods (see Liebmann et al., 2018). As VOCs in this study were sampled at the same location as the total OH reactivity, the effect of inhomogeneity of the air composition is minimized. However, the ozone mixing ratio used to derive the ozone correction (described in Sect. 2.5.1) is retrieved from the station mast (115 m away) and at a height of 4.2 m. It is very likely that emissions from soil and understorey vegetation (or from standing water close to the OH reactivity sampling location) would further deplete the ozone close to the ground, leading to an overestimation of the correction. Under some circumstances, such as when there is a strong O3 gradient below canopy (Chen et al., 2018), the correction might be overestimated.
For instance, on 29 and 30 April total OH reactivity around 125 to 150 s−1 in the afternoon is followed by O3 concentration drops below canopy (Fig. 4; see also Chen et al., 2018). While the high reactivity peaks themselves are likely not affected by an overestimation of the correction, the period following them (night-time) might be slightly overestimated due to the sampling of O3 further away and higher above ground. This effect is difficult to take into account in retrospect. The concentration of O3 should have been measured immediately next to the CRM system. Similar conditions were observed during nights between 11 and 16 May and to some extent in July (without reaching such high total OH reactivity values as in spring). This effect on the inhomogeneity of the forest air composition might affect total OH reactivity measurements and in turn partly explain some of the missing fraction.
2.5.2 1st-order correction factor
Sinha et al. (2008) used a two-equation model to correct for the deviation from pseudo-1st-order kinetics ([Pyr]≫[OH]). Michoud et al. (2015) used more detailed modelling taking into account OH recycling reactions, but could not match the model results with their experimental data. For this reason, Michoud et al. (2015) favoured the experimental approach to correct the reactivity data. Nevertheless, the experimental approach also has drawbacks. For instance, impurities from standards and changes over time (ageing) might alter its reactivity. Also it is based on calibrations using one compound at a time, which do not represent complex ambient mixtures of reactive gases.
Nevertheless, reactivity calibrations were performed for the present study with a 10 ppmv C3H8 standard as well as with an in-house gas mixture containing α-pinene with small impurities from aromatic compounds. The concentrations of the C3H8 and in-house α-pinene standards were checked periodically by taking adsorbent tube samples and analysing them by GC–MS. At the same time impurities (4.7 %–17 % of the reactivity) could be measured and taken into account.
The comparison between OH reactivity expected from the standard (Rstd) and the measured OH reactivity (Rmeasured) is presented in Fig. 5. Note that the data have been corrected for deviation from pseudo-1st-order kinetics similarly to the work of Sinha et al. (2008) by using numerical simulations and fitting the relationship between Reqn and Rtrue for various pyr ∕ OH ratios with equations of the form , so that F1, F2, and F3 are pyr ∕ OH-dependent coefficients (see Appendix A for additional details).

Figure 4The 1 h averages of total measured OH reactivity, Rexp, and ozone mixing ratios at 4.2 and 125.0 m above ground. Mixing layer height (MLH) is shown as a grey shadow. Note that the detection limit for MLH is 60 m and values below this limit are displayed at 30 m (and zeros denote gaps in the data).
The calibration for C3H8 is consistent with the one from Sinha et al. (2008). Due to the high reactivity of α-pinene, the calibration consistently underestimates the expected reactivity and because monoterpenes constitute the most important class of compounds in the boreal forest, this needs to be taken into account by applying the overall correction for α-pinene to the ambient data in this study, which has an uncertainty (UF) of 6.3 % based on the uncertainty of the fit. The reactivity measured in the reactor Rmeasured is then derived from the following equation:
Based on these reactivity calibrations the precision of the measurements (Uprec) is derived. Reqn and its standard deviation () are calculated for every C3 value measured. Dividing by the mean of Reqn () for stable conditions yields Uprec, which varies with Reqn values and is described by the following function:
This derivation is shown in Figure B1 of the Appendix.
2.6 The model to Simulate the concentrations of Organic vapours, Sulfuric Acid and Aerosols (SOSAA)
In this study we applied the model to Simulate the concentrations of Organic vapours, Sulfuric Acid and Aerosols (SOSAA) to simulate the OH reactivity at the SMEAR II station for selected days in April, May, and July 2016. SOSAA is a one-dimensional chemical transport model comprised of boundary layer meteorology, biogenic emission of VOCs, gas-phase chemistry, aerosol dynamics, and gas dry deposition (e.g. Boy et al., 2011; Zhou et al., 2014), and it has been previously used to simulate OH reactivity at this site (Mogensen et al., 2011, 2015).
The boundary layer meteorology was derived from scalar distribution (SCADIS; Sogachev et al., 2002), as described in Boy et al. (2011). The biogenic emission module was deactivated because in situ measurements were used to provide input concentrations. Biogenic compounds were set to the measured values up to 18 m (canopy height), while aromatic compounds were set to the measured values at all heights. Measured inorganic gas concentrations at SMEAR II were used as input.
The gas-phase chemistry was created using the Kinetic PreProcessor (KPP; Damian et al., 2002). The chemical reaction equations used in this study were selected from the Master Chemical Mechanism v3.3.1 (MCMv3.3.1 Jenkin et al., 1997, 2012, 2015; Saunders et al., 2003; Bloss et al., 2005). The chemistry scheme included more than 15000 reactions and a total of 3525 chemical species representing the complete reaction paths for isoprene, α-pinene, β-pinene, limonene, β-caryophyllene, methane, 2-methyl-3-buten-2-ol (MBO), benzene, toluene, styrene, ethylbenzene, 1,2-dimethylbenzene, 1,3-dimethylbenzene, 1,4-dimethylbenzene, 1,2,3-trimethylbenzene, 1,2,4-trimethylbenzene, 1,3,5-trimethylbenzene, 1-ethyl-2-methylbenzene, 1-ethyl-3-methylbenzene, 1-ethyl-4-methylbenzene, heptane, octane, nonane, butanal, pentanal, methacrolein, and relevant inorganic reactions. The 1st-order reactions between OH, O3, and NO3 with the following monoterpenes were also included in the chemistry: Δ3-carene, myrcene, camphene, and 1,8-cineole. Likewise, 1st-order reactions between OH, O3, NO3, and β-farnesene were included. The photochemistry has been improved by calculating the photodissociation constants more precisely using data from Atkinson et al. (1992), as described in Mogensen et al. (2011). The OH reactivity has been calculated as in Mogensen et al. (2011, 2015). The condensation sinks for sulfuric acid and nitric acid, based on differential mobility particle sizer (DMPS) and aerodynamic particle sizer (APS) data from SMEAR II, were included (Boy et al., 2003). Since sulfuric acid and nitric acid make up most of the condensation sinks, sinks of VOCs into the particle phase are not taken into account, thereby the aerosol module is turned off.
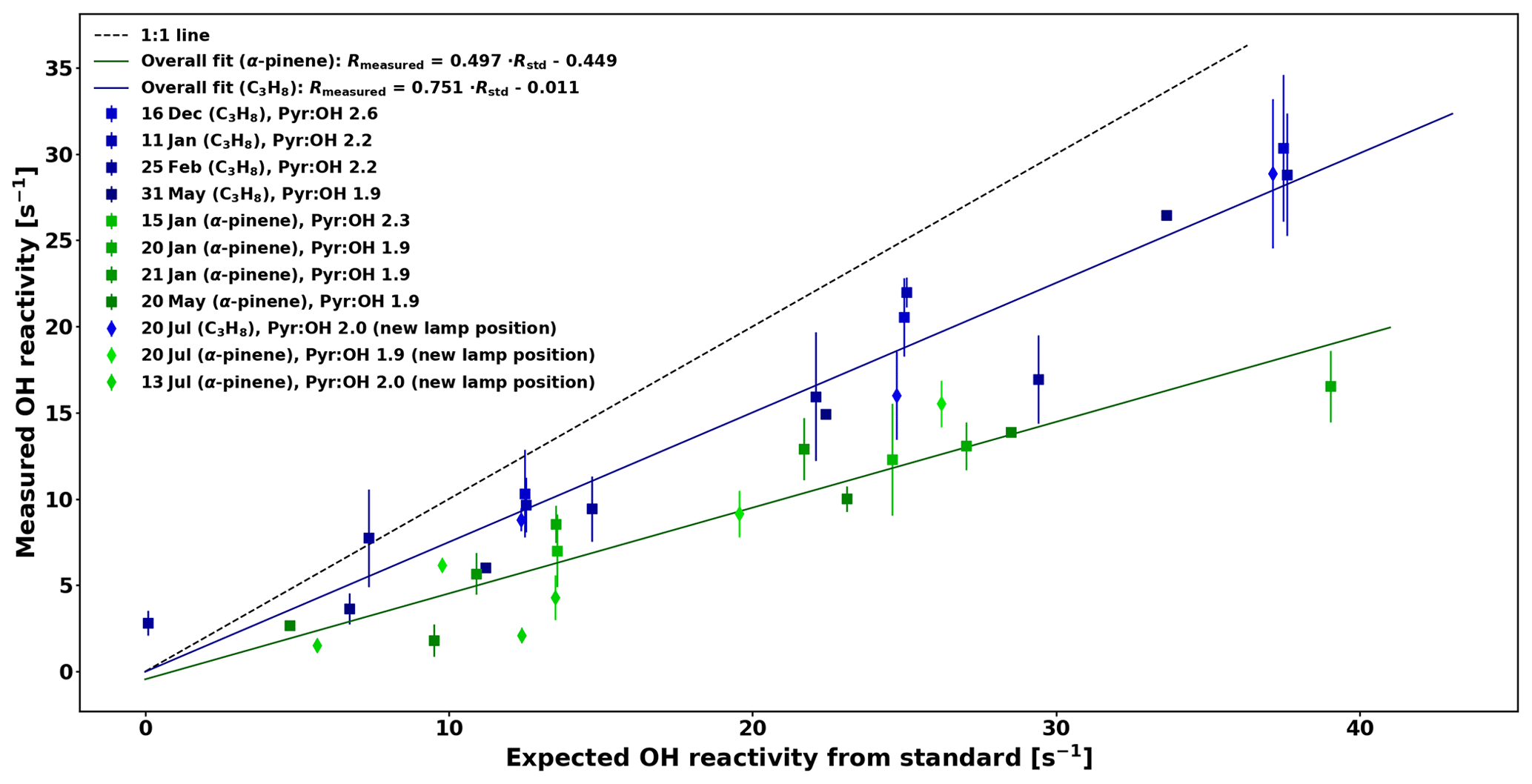
Figure 5Comparison between measured OH reactivity for C3H8 and α-pinene standards with the expected OH reactivity.
The model runs in the present study include the dry deposition module implemented in SOSAA by Zhou et al. (2017a) and extended in Zhou et al. (2017b). The latter describes the explicit simulation of the loss of every compound in the model by dry deposition inside the canopy for all height levels and provides a detailed comparison of measured and modelled fluxes of certain selected VOCs including some secondary organic species at SMEAR II.
3.1 Overview
An overview of the measured total OH reactivity together with the calculated OH reactivity from up to 104 compounds, depending on data availability, as well as selected ancillary data, such as environmental conditions (air and surface soil temperatures as well as surface soil water content), and contributions from different compounds and groups of compounds are presented in Fig. 6. The following sections discuss in detail various aspects of the results such as (a) seasonality, (b) diurnal variations, and (c) missing reactivity. Nevertheless, from this overview, the following observations can be made.
-
The range of measured total OH reactivity values is similar to that of previous studies at the same site in August 2008 and July–August 2010 (Sinha et al., 2010; Nölscher et al., 2012), with notably higher values in the spring.
-
These high total OH reactivity peaks in the spring (with values higher than at the end of July) seem to be associated with changes in the soil water content resulting from soil thawing.
-
The calculated OH reactivity from measured compounds is in general lower than the measured total OH reactivity (also for periods with a large number of compounds included in the analysis), leading to a large fraction of missing reactivity (see Sect. 3.4).
-
Inorganic compounds (CH4, CO, O3, and NO2) form an important fraction of the calculated OH reactivity.

Figure 6(a) Experimental total OH reactivity Rexp (1 h average) and calculated OH reactivity ROH, (b) environmental conditions (air and surface soil temperatures, as well as surface soil water content), (c) Pyr ∕ OH in the CRM reactor, (d) data availability from the different instrumentation/sources, (e) fraction of experimental total OH reactivity, and (f) fraction of calculated OH reactivity. The periods shaded in grey in (a) to (d) represent the periods investigated with SOSAA (see Sect. 3.4).
3.2 Total OH reactivity
Table 1Monthly means and standard deviations (SD) of experimental total OH reactivity (Rexp), the missing OH reactivity fraction (Rmissing,fraction), monoterpene and sesquiterpene mixing ratios ([MT] and [SQT], respectively), photosynthetically active radiation (PAR), precipitation (Precip), relative humidity (RH), air temperature (T), surface soil temperature (Tsoil,humus), surface soil water content (wsoil,humus), and mixing layer height (MLH). Coefficients a and b from linear regressions between the weekly means of these variables and weekly averaged Rexp and the corresponding coefficients of determination (r2). ndays indicates the number of days with measurements. n denotes the number of Rexp observations. Note that all other means (except MLH) have been derived for the same measurement period as Rexp. nMLH indicates the number of observations with overlapping Rexp and MLH measurements.
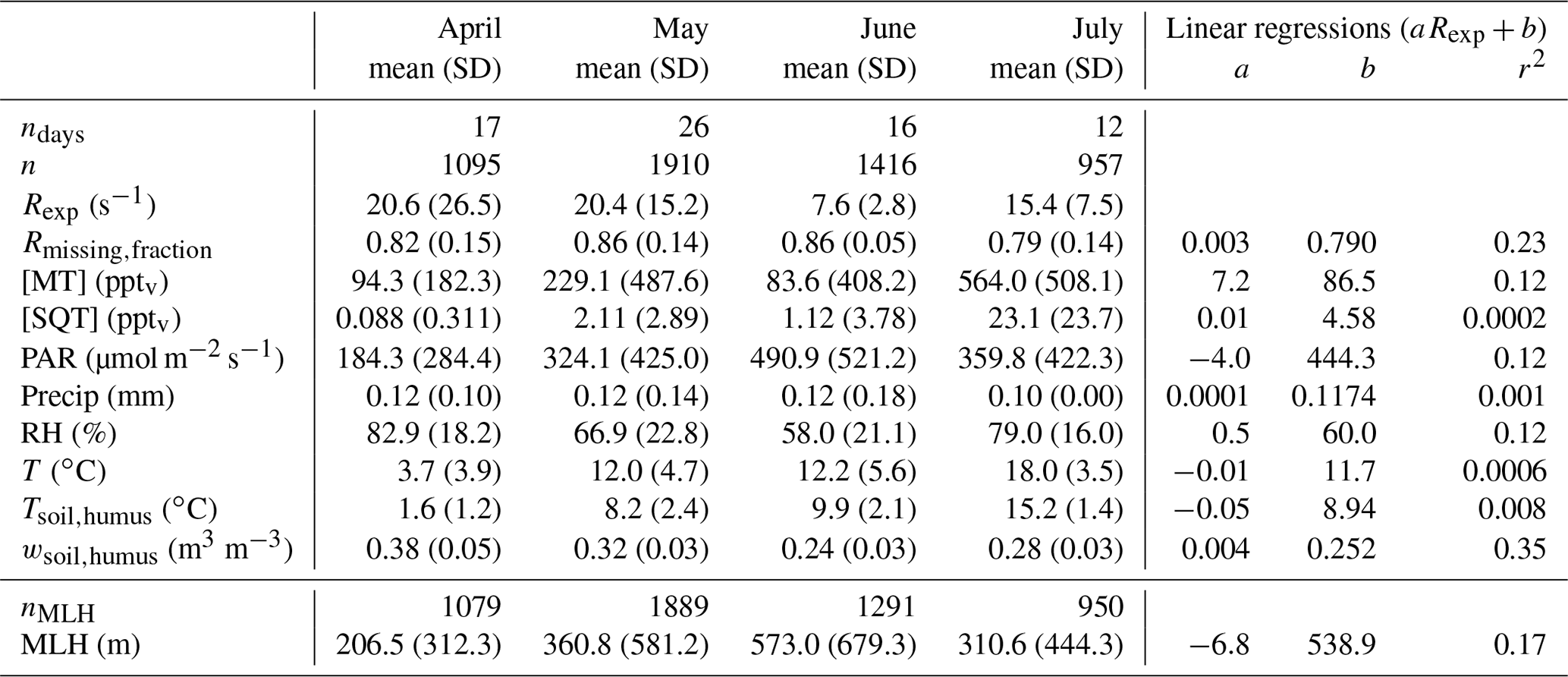
Keeping in mind that the experimental data have not always been acquired continuously, the total experimental OH reactivity (Rexp) monthly mean was high in April and May (about 20 s−1) compared to June (7.6 s−1) and July (15.4 s−1), due to few very high values at night-time (Table 1). Consequently, no strong correlation could be found between Rexp and other variables looking at weekly means. The highest coefficient of determination (r2) was obtained for the correlation with wsoil,humus (r2 = 0.35), which indicates that soil moisture might be an important driver for the high reactivity values measured in spring. The highest reactivity peaks happened when the surface soil water content was the highest as the surface soil temperature started to increase above 1.5 ∘C, indicating thawing of the soil, a possible source of OH reactive compounds. Forest floor emissions of monoterpenes are known to be high in spring after snow has melted (Hellén et al., 2006; Aaltonen et al., 2011; Mäki et al., 2017), and VOC emission bursts have been observed after wetting events (e.g. Rossabi et al., 2018). There has also been some indication that thawing snow/soil could be a source of volatile organic amines (Hemmilä et al., 2018). In the present study, the soil was snow-free already on 8 April, but a short snowfall episode happened later with 5 cm of snow measured on the morning of 25 April (which was gone on the next day). This episode happens just before the first OH reaction peak (at about 56 s−1), but this single occurrence is too little information to reach conclusions about the role of snow in the large OH reactivity values observed, and it might well be due to a combination of factors (including snowfall and immediate melting). These results deviate however from the conclusions of Nölscher et al. (2016), which suggested that a wet (and cold) soil in the Amazon rainforest acts as a sink for reactive compounds.
The data for July cover days that were cloudier and more humid (both air and soil) but warmer than the period covered by the data in June leading to higher total OH reactivity. Monthly means of ambient concentrations of locally emitted terpenoids had a weak correlation (r2=0.43) with temperature (see also Hellén et al., 2018), which is not reflected in the correlation of total measured OH reactivity with temperature, as observed earlier (e.g. Nakashima et al., 2014; Ramasamy et al., 2016). However, these studies were performed during summer, which highlights the different regimes governing OH reactivity in various seasons and how most likely other (unknown) compounds in addition to terpenes contribute to OH reactivity during spring. In other words, while conditions that favour high OH reactivity values seem to favour BVOC (terpene) emissions in the summer as well, OH reactivity is driven by other parameters in spring.
It should be noted, though, that the use of a correction factor based on α-pinene throughout the measurement period even though the air composition varied might lead to an overestimation of the measured total OH reactivity. However, average mixing ratios of monoterpenes were similar in April and June (94.0 and 83.6 pptv, respectively), so that relative differences in measured total OH reactivity cannot be explained this way. This further indicates that non-terpene compounds that were not measured in the spring might have contributed to the total OH reactivity.
3.3 Diurnal variations
The calculated OH reactivity of various groups of compounds shows different diurnal patterns, which vary with the season as well. Their normalized values are depicted in Fig. 7 (second to fourth row), separated by month (April to July in columns), together with the normalized diurnal patterns of Rexp and its missing fraction and temperature difference between measurements at 4.2 m and 125.0 m above ground as a proxy for mixing layer height (top row). Compounds that had a 24 h sampling time were removed from this analysis. Sinha et al. (2010) did not measure a clear OH reactivity diurnal pattern during their 2-week measurement period and the modelling of the OH reactivity also showed no diurnal pattern (Mogensen et al., 2011). However, Mogensen et al. (2015) modelled a weak diurnal pattern with a maximum at night, mostly due to improvements in the meteorological scheme. The observations in the present study, even though at higher OH reactivity levels, show this pattern from May to July. Nölscher et al. (2012), for measurements roughly at the same time of the year, identified a similar diurnal pattern with a maximum at night during the identified stress period. For normal boreal forest conditions, they measured large variations in the afternoon reactivity, sometimes leading to a maximum, which they associated with long-range transport. In the present study, afternoon reactivity maxima were dominating April's diurnal pattern.
When the total measured OH reactivity hourly average is at a minimum during the day and a maximum at night (May to July), it follows the pattern of BVOC concentrations (and calculated OH reactivity) due to the low mixing layer height and despite slightly lower emissions due to the lower temperatures at night (Hellén et al., 2018). The hourly average of missing reactivity fraction remained consistently high (between 71 % and 92 %), similar to values from Nölscher et al. (2012) and despite the inclusion of more compounds in our analysis (see Sect. 3.4 for a detailed discussion).
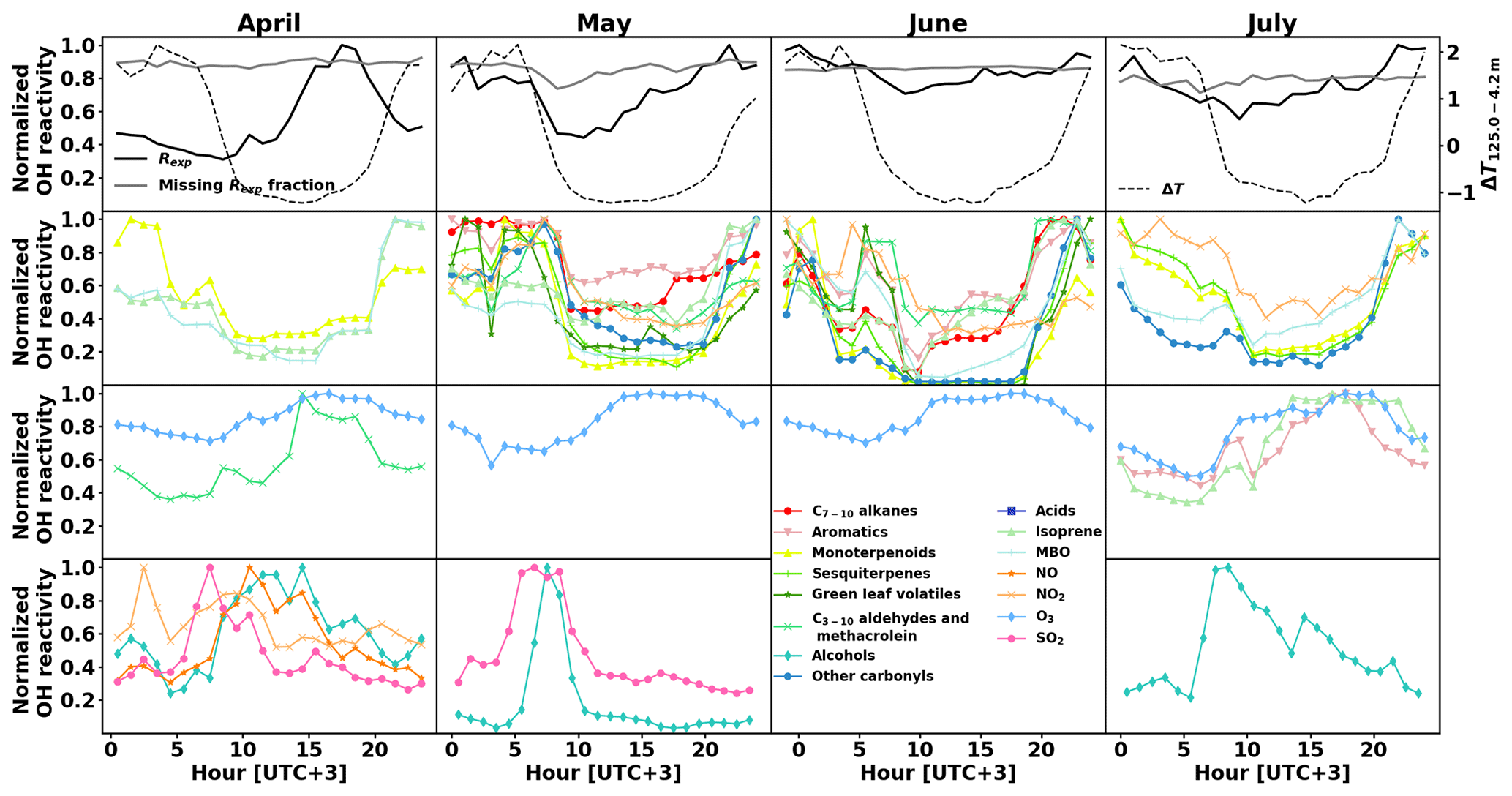
Figure 7Normalized monthly averaged diurnal variations in experimental OH reactivity Rexp and the missing fraction as well as temperature gradient between 4.2 and 125.0 m above ground as a proxy for mixing layer height (top row), and calculated OH reactivity separated by group of compounds (second to fourth rows).
While the OH reactivity daily patterns from monoterpenoids and MBO had a minimum during the day for all months, other groups of compounds showed this reactivity pattern only for some periods. Isoprene showed this pattern except in July, where the light-induced emissions during the day dominated. Sesquiterpenes, other carbonyls, and NO2 showed a similar pattern with a daytime minima from May to July, while C7−10 alkenes, aromatics, C3−10 aldehydes, and methacrolein showed a pattern with daytime minimum only in May and June. Alcohols exhibit an OH reactivity pattern with a maximum in the morning (09:00–11:00). The absolute OH reactivity of alcohols is low and dominated by 1-butanol, which is used in aerosol measuring devices at the site. It is not clear what causes the diurnal pattern, but SO2 reactivity had a similar pattern in April and May, and NOx had such a pattern in April, when the photochemistry is not yet very strong.
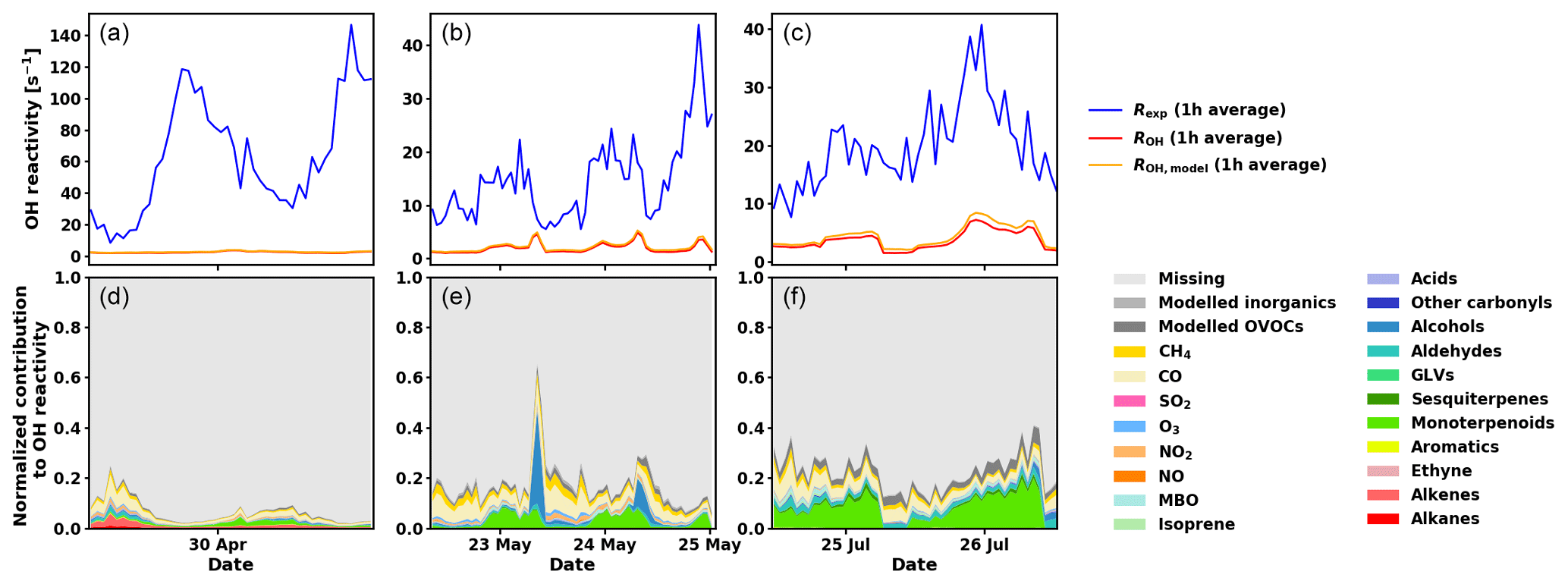
Figure 8Measured total OH reactivity (Rexp), calculated OH reactivity from measured compounds (ROH), calculated OH reactivity including measured and modelled compounds (ROH,model) (1 h averages, a, b, c), and normalized contributions to Rexp for various compounds and groups of compounds (d, e, f) for the three periods investigated with SOSAA (see main text for details).
Overall, from May to July the total OH reactivity exhibits a minimum during the day and a maximum at night, following the OH reactivity pattern for biogenic compounds (except for isoprene in July, which is present in low concentrations in this pine forest, and has a maximum in the afternoon then). In April, the total OH reactivity has a maximum in the afternoon, and sesquiterpenes, even though present in low concentrations, show a similar reactivity pattern. Mäki et al. (2019) found high levels of sesquiterpenes from soil emissions at the same site in spring. This is an additional indication that unknown primary emissions (in particular from soil) could drive the reactivity during that time of the year.
3.4 Missing OH reactivity
The comparison between the calculated and measured OH reactivity is challenging as the calculated values are derived from a number of compounds that varies because of the availability of the measurements (Fig. 6d). Some periods include only a few inorganic compounds from the station mast while other periods include a large number of (O)VOCs analysed by the GC–MSs. The contribution to the known reactivity is shown in Fig. 6f. It is also good to keep in mind that part of the missing reactivity can be explained by measurement uncertainties and potential overestimation due to applied correction factors. As the data in this study have been uniformly corrected based on α-pinene calibrations, it is likely that the obtained values are an upper limit for the reactivity considering that α-pinene (and monoterpenes in general) do not dominate the air composition and reactivity at the site for the whole measurement period. It should also be remembered that because of technical problems, PTR-MS data (VOC data) from the station mast are unavailable for our measurement period. Some compounds such as acetaldehyde were measured during two short periods with offline 24 h sampling methods. In late July, acetaldehyde contributed on average 0.13 s−1 to the OH reactivity, which can be a small but significant fraction for low OH reactivity values. This is likely the case for other compounds that were not measured at all in the present study such as formaldehyde, acetonitrile, or methanol, to name a few. In addition, this also makes the comparison with previous studies difficult. Despite the higher number of compounds included in the present work, the impossibility to include aforementioned compounds in the analysis partly explains why missing OH reactivity fractions remain high. Therefore, even with the maximum number of compounds used to calculate OH reactivity (late April–early May) a large fraction of the measured total OH reactivity remains unexplained (missing reactivity, Fig. 6e).
However, this period also coincided with high reactivity peaks observed likely due to soil thawing as mentioned previously. Only sesquiterpenes peaked at the same time as the total OH reactivity, but their concentrations are still low, which is why we mentioned amines and non-terpene BVOCs as potential classes of compounds contributing to the observed total OH reactivity. Kumar et al. (2018) identified various non-hydrocarbon classes of compounds associated with biomass burning that potentially contribute to OH reactivity. However, even if long-range-transported biomass burning emissions are observed occasionally at the measurement site of this study (Leino et al., 2014), no significant increase in CO concentrations above 250 ppbv was observed during the measurement period as in Leino et al. (2014). Only between 23 and 26 July were concentrations of 150 ppbv (slightly above the average background levels of 100 ppbv) detected. Nevertheless, these classes of compounds could potentially be emitted by local sources of a different kind.
As has been shown for forests dominated by isoprene emitters (Kim et al., 2011; Kaiser et al., 2016), oxidation products from BVOCs might contribute significantly to the missing OH reactivity. As oxidation products of monoterpenes and sesquiterpenes are neither measured routinely nor were they monitored for this study, the SOSAA model was used (see Sect. 2.6) using measured trace gases and meteorological conditions as inputs in order to calculate the potential contribution of terpene oxidation products to missing OH reactivity. Three periods of 2 to 3 d for the months of April, May, and July were simulated. The results for the inclusion of modelled oxidation compounds in the analysis are presented in Fig. 8. These compounds labelled modelled OVOCs are mostly peroxides, alcohols, and carbonyl compounds due to the generally low NOx levels at the site. Modelled inorganics, whose contributions are negligible, regroup molecular hydrogen (H2), hydrogen peroxide (H2O2), nitrous acid (HONO), peroxynitric acid (HO2NO2), nitric acid (HNO3), and the nitrate radical (NO3). To check the model performance for the concentrations of the secondary organic species, we compared nopinone measured by GC–MS with the model output. The plots for all three selected periods (Fig. C1 in the Appendix) show that the trend of the model concentrations for nopinone are comparable to the measured values and the absolute values are mostly inside estimated 50 % uncertainties of the measurements. The only exceptions are the difference during daytime in May when the measurements show a very strong decrease in the morning, but the model follows this behaviour much more weakly. No other specific secondary species is available for such comparison due to the lack of measurements.
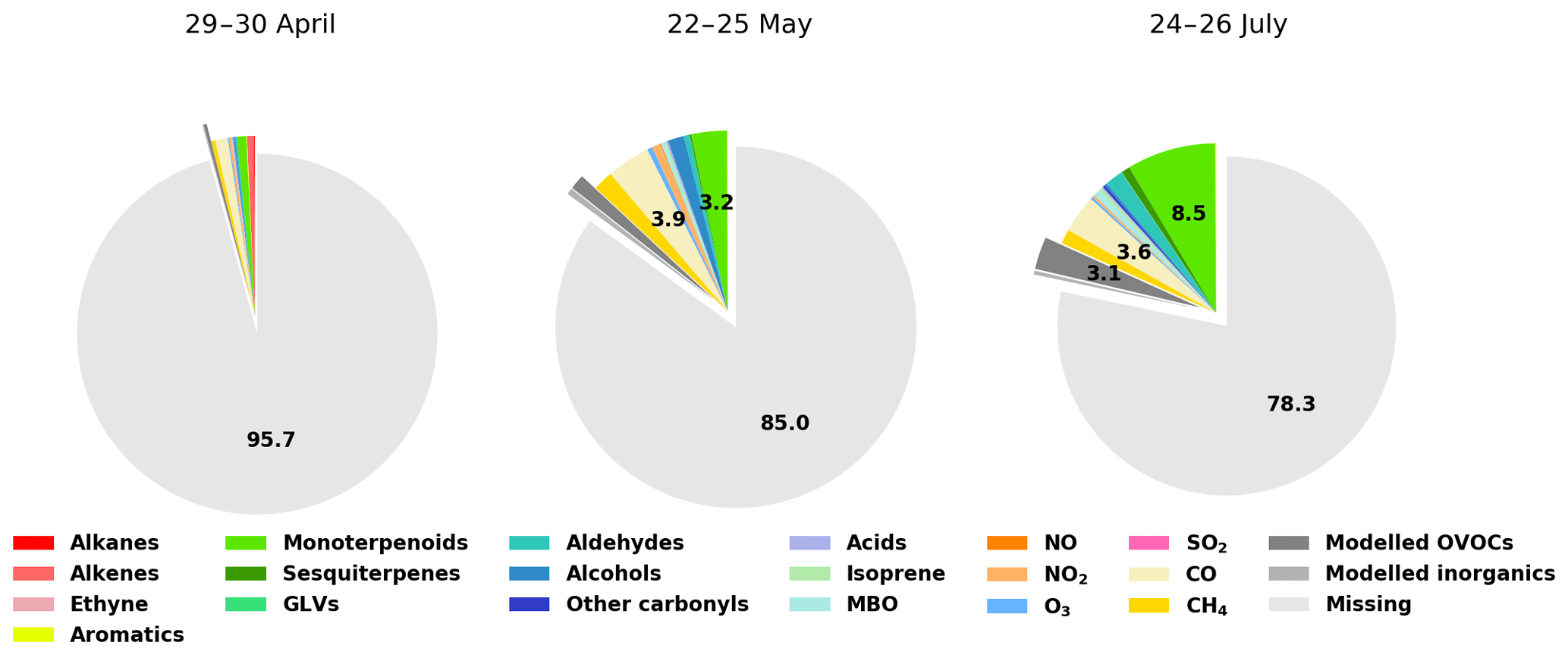
Figure 9Contributions of various compounds and groups of compounds to the measured total OH reactivity (Rexp). For clarity, labels for fractions smaller than 2.0 % have been omitted.
While the trend of ROH,model qualitatively follows the general trend of Rexp, ROH,model underestimates Rexp, especially at night. Total OH reactivity values are in general lower during the day and they are closer to ROH,model values. In late April and early May, the high peaks in the late afternoon indicate missing primary emissions, which also contribute (or their oxidation products) to the missing reactivity in the following nights.
Retrieving the additional reactivity from these modelled compounds that were not included in ROH reduced the missing reactivity by only a small fraction (about 4.5 % for the studied period in July and less for the other periods) as seen in Fig. 9. A detailed breakdown of the individual compounds contributing to the reactivity and their mixing ratios can be found in Appendix D.
Most of the missing reactivity could then be due to oxidation products that are not included in the model from measured precursors such as Δ3-carene, myrcene, camphene, 1,8-cineol, β-farnesene, or unidentified sesquiterpenes (in contrast with the well-studied isoprene chemistry scheme), but the contribution to the OH reactivity from these precursors is small due to their low atmospheric concentrations, so that the contribution from their oxidation products is also expected to be small (Hellén et al., 2018). As mentioned earlier, missing primary emissions also contribute to the missing reactivity, more so in spring than in summer.
Amines released from soil, as mentioned previously, are a potential class of compounds that could contribute to OH reactivity. Kumar et al. (2018) identified various non-hydrocarbon classes of compounds associated with biomass burning that potentially contribute to OH reactivity.
It is also good to keep in mind that part of the missing reactivity can be explained by measurement uncertainties and potential overestimation due to applied correction factors. As the data in this study have been uniformly corrected based on α-pinene calibrations, it is likely that the obtained values are an upper limit for the reactivity considering that α-pinene (and monoterpenes in general) do not dominate the air composition and reactivity at the site all the time.
A previous study by Mogensen et al. (2011) modelled the OH reactivity at the SMEAR II site for the year 2008, using modelled emissions, and estimated the OH reactivity to be about 2–3 s−1 between April and July. This is lower than the measured averages from the present and previous studies and lower than the night-time modelled values in July from the present study. Mogensen et al. (2011) report that secondary organics, β-caryophyllene, farnesene, and MBO represent 8 % of the total OH reactivity, which represents the same magnitude as the results from this study. Mogensen et al. (2015) modelled the OH reactivity at the same site for July and August 2010 with the same methodology (including minor model improvements) and obtained values between 2.7 and 3.2 s−1. The higher modelled values in our study indicate that modelled emissions lead to lower monoterpene concentrations than measured concentrations.
Our results are not entirely in line with other studies that showed reductions of the missing reactivity by constraining VOC concentrations to model their oxidation products (e.g. Mao et al., 2012; Edwards et al., 2013; Kaiser et al., 2016), as the reduction observed remains small in this study. This approach still leaves a large unexplained fraction of OH reactivity. This is a strong indication that, on the one hand, non-terpenoid compounds contribute to the total OH reactivity and that, on the other hand, more compounds have to be included in the chemical model.
Finally, heterogeneous loss of OH to particles might be a contribution to missing OH reactivity, but this process is poorly quantified (Donahue et al., 2012). Due to the low sampling flow and long FEP sampling line to the CRM instrument, it is unlikely that particles will reach the reactor. Additionally, we could not find any correlation between ambient particle numbers and either total measured OH reactivity or its missing fraction.
As a side note, total OH reactivity measurements were unfortunately not available in the autumn, but Liebmann et al. (2018), who measured nitrate radical (NO3) reactivity at the same site, found about 30 % of unexplained NO3 reactivity at night and about 60 % during daytime. Mogensen et al. (2015) modelled NO3 reactivity at the site and found a maximum in the early morning, while the measurements from Liebmann et al. (2018) showed a maximum at night. The modelled NO3 reactivity values were similar to the measured ones without strong temperature inversion at night, while higher measured values were recorded for nights with strong temperature inversion.
Hellén et al. (2018) showed that the balance between the emissions of VOCs and the production of oxidation compounds and the sinks varies with the season, leading to different diurnal profiles for compounds such as isoprene, C7−10 aldehydes, and nopinone. This can also be observed in terms of OH reactivity in the present study (see Sect. 3.3).
Total OH reactivity is not a simple function of a few variables. It includes many complex processes involving sources and sinks that can change dramatically depending on the environmental conditions and the time of the year. Measurement uncertainties and data availability for comparison between measured total OH reactivity values and calculated values also represent a challenge when interpreting results.
In the present study total OH reactivity measurements were performed at a Finnish boreal forest research site (SMEAR II). The monthly averaged experimental total OH reactivity was high in April and May (about 20 s−1) due to some very large afternoon reactivity peaks captured when the soil was thawing. The low sampling height and the peaking of sesquiterpene emissions, which are known to be emitted from soil, at the same time as OH reactivity in April indicate that the forest floor is a potential important but overlooked source of reactive compounds. The total OH reactivity diurnal pattern from May to July follows that of biogenic compounds with high values during the night due to the low mixing height, even though emissions are lower at night.
A suite of online and offline (O)VOCs measurements was used to calculate the known fraction of OH reactivity to compare it to the total OH reactivity measured. The missing fraction of the OH reactivity remained high for the measurement period. This can be due to various reasons. As the data availability of (O)VOCs varies, the comparison between experimental and calculated OH reactivity is difficult but three different explanations can lead to high missing (unexplained) OH reactivity: (1) simply the lack of measurements, (2) not measuring oxidation products (only their precursors), and (3) not measuring the right class of compounds. We showed that compounds not included (or only partially included) in the analysis due to the unavailability of measurements (e.g. due to technical problems), such as acetaldehyde, might contribute a small but significant fraction to the total OH reactivity, in particular for low reactivity values. Using a one-dimensional transport model to estimate oxidation product concentrations from measured precursor concentrations for three short periods of 2 to 3 d in various months (with most (O)VOC data availability) it is demonstrated that only a small fraction (up to ca. 4.5 %) of the missing reactivity can be explained by these oxidation products. On the one hand, this is due to the absence in the model of a degradation scheme for detected compounds in the ambient air (e.g. Δ3-carene, β-farnesene), but on the other hand it is also possible that non-hydrocarbon compounds contribute to the OH reactivity as well.
More measurements of oxidized compounds and identification of non-terpene reactive compounds from emissions from sources other than vegetation (e.g. soil) are required to better understand the reactivity and local atmospheric chemistry in the forest air in general, in particular during winter, spring, and autumn, when the forest air chemistry is not dominated by emissions from the vegetation.
All the data whose source is not explicitly specified in the text are available upon reasonable request to the corresponding author.
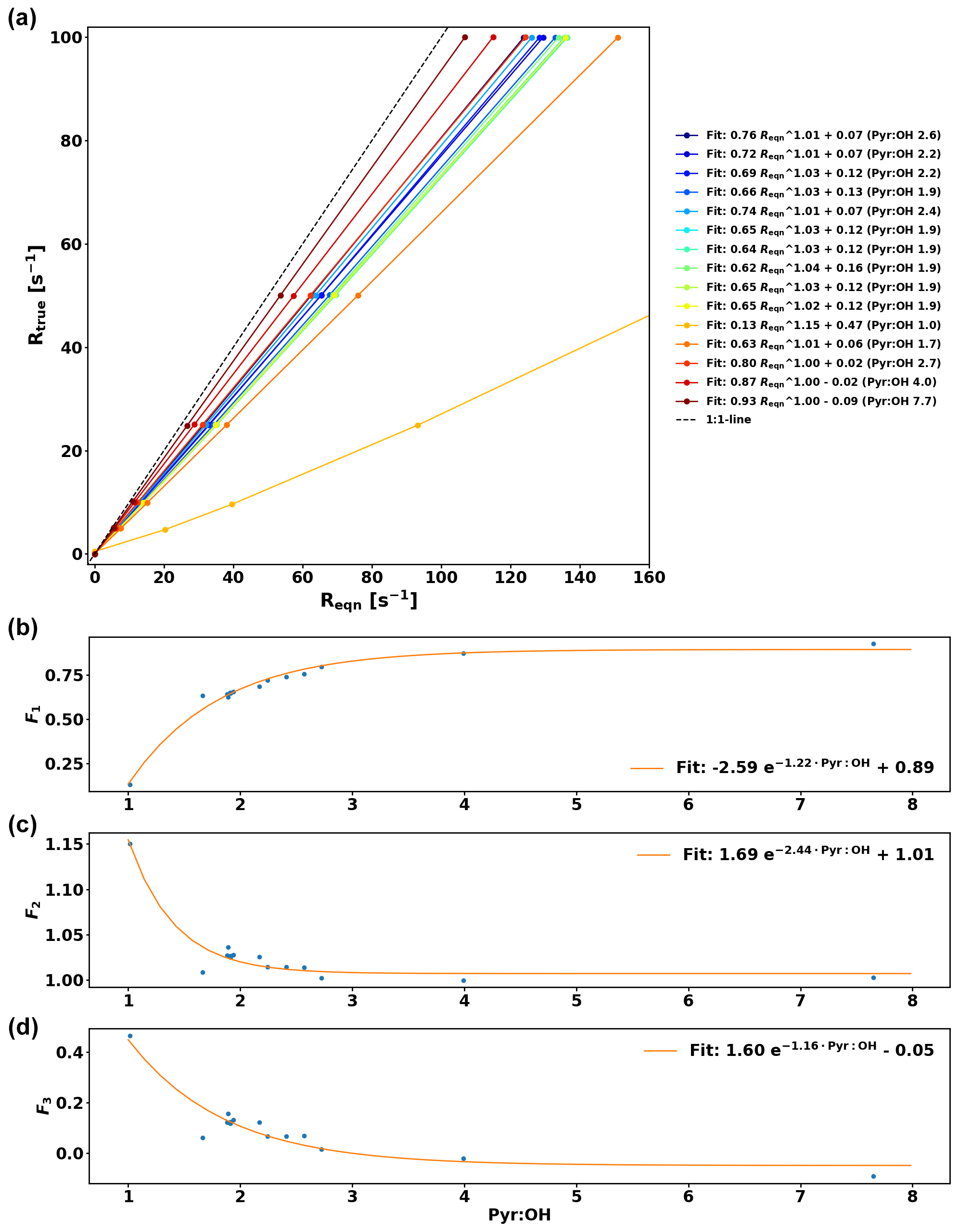
Figure A1Numerical simulations of Rtrue as a function of Reqn for various pyr ∕ OH values and their corresponding fit curves of the form (a). Fit coefficients F1, F2, and F3 as a function of pyr ∕ OH and corresponding exponential fit curves (b, c, d).
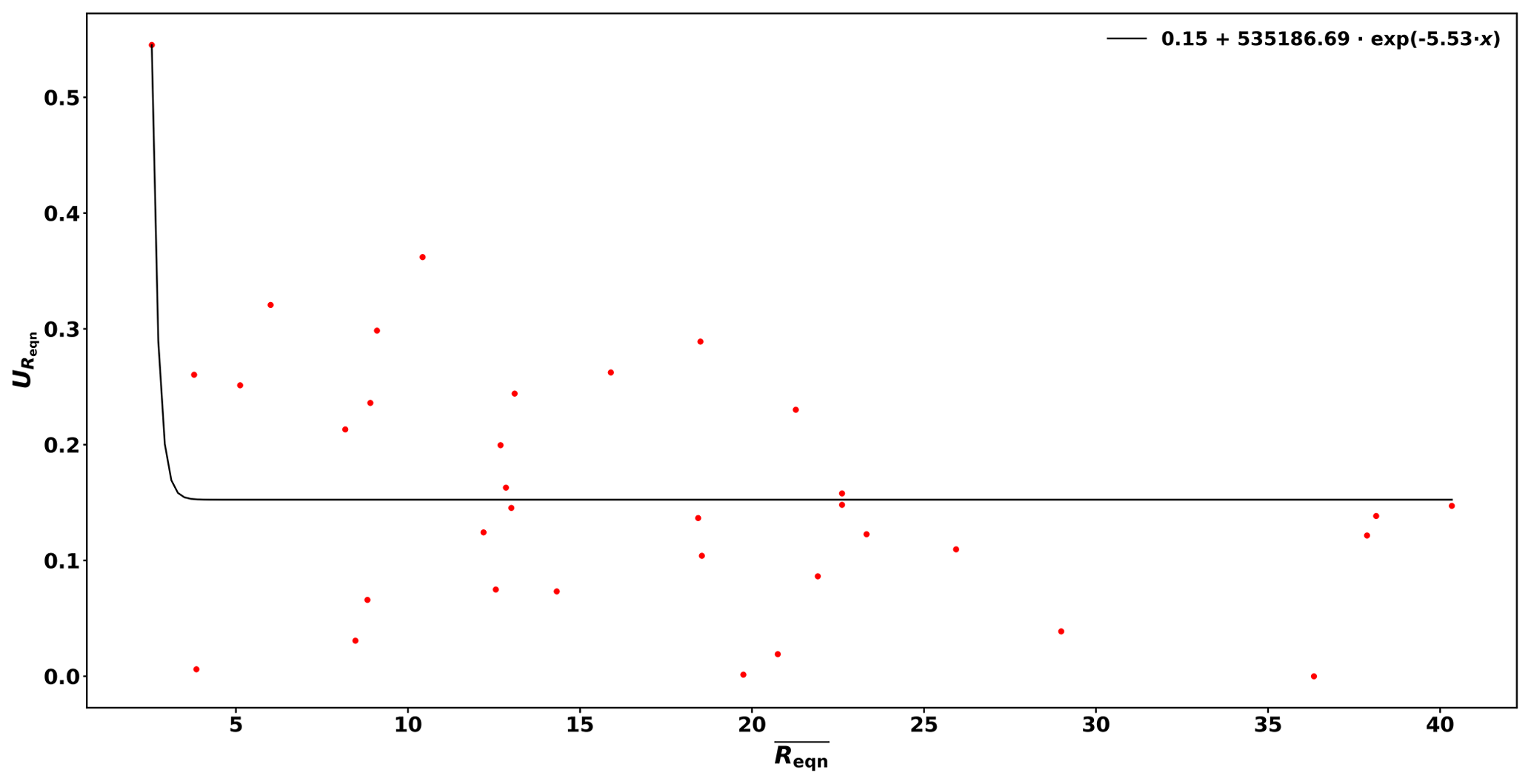
Figure B1Uncertainty of Reqn () as a function of . The solid line is the fit function for the precision of the measurements (Uprec).
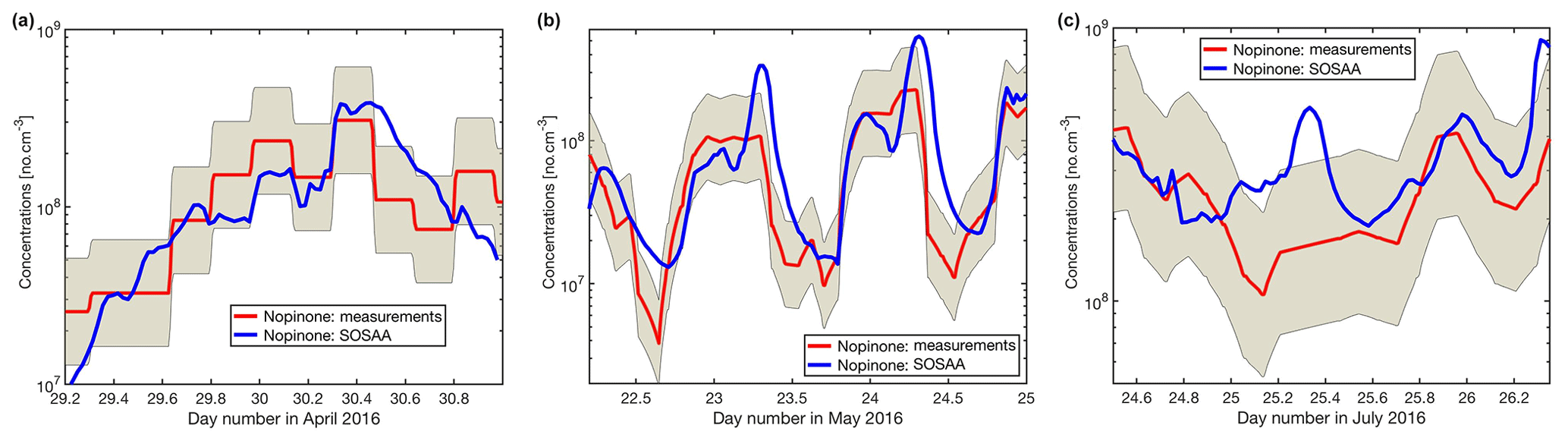
Figure C1Mixing ratio of measured (red line and shaded area for 50 % uncertainty) and modelled (blue line) nopinone for the three modelled periods in April, May, and July (a–c).
Table D1Averages of individual compound mixing ratios (pptv) and calculated OH reactivity, ROH (s−1), and group ROH for the three periods studied with SOSAA. “n.d.” means “not detected” and “n.m.” means “not measured”.
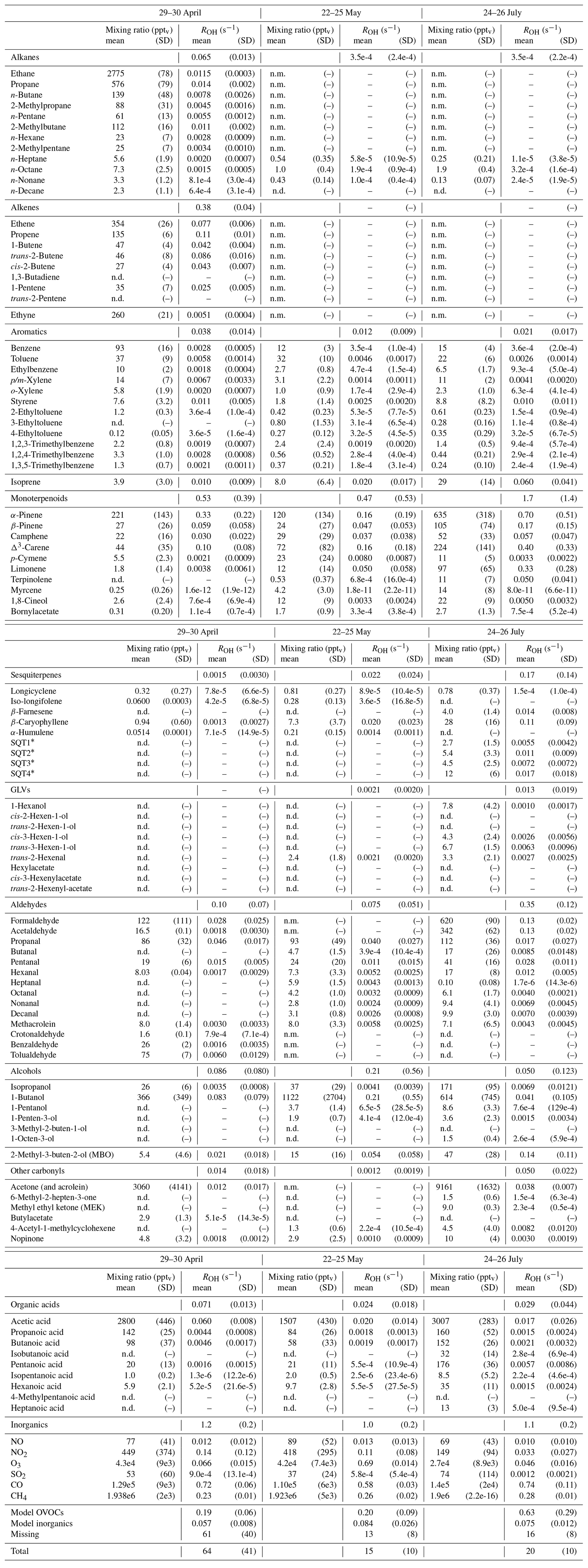
∗ Quantified with β-caryophyllene calibration and an estimated reaction coefficient (1e-10 cm3 s−1).
APP conducted total OH reactivity measurements, offline sampling, and LC–UV analysis; performed data analysis; and led the writing of the paper. HH designed the measurement campaign, conducted GC–MS measurements and data analysis, and commented on the paper. TT assisted with GC–MS measurements and data analysis and commented on the paper. VV provided mixing layer height data and their description in the method part and commented on the paper. DC, MB, and PZ performed model runs with the help of DT and all commented on the paper.
The authors declare that they have no conflict of interest.
The authors thank Hannele Hakola for the continuous support. They also thank the staff at the SMEAR II station for their help, Petri Keronen for providing the data that we retrieved from SmartSMEAR, Jari Waldén for lending calibration standards and gas analysers, and Anne-Mari Mäkelä for the analysis of the canister samples. The authors also wish to acknowledge the CSC IT Center for Science, Finland, for computational resources.
This research has been supported by the Academy of Finland (grant no. 275608), the Academy of Finland, Biotieteiden ja Ympäristön Tutkimuksen Toimikunta (grant no. 307957), and the Academy of Finland (grant no. 272041).
This paper was edited by Yugo Kanaya and reviewed by two anonymous referees.
Aaltonen, H., Pumpanen, J., Pihlatie, M., Hakola, H., Hellén, H., Kulmala, L., Vesala, T., and Bäck, J.: Boreal pine forest floor biogenic volatile organic compound emissions peak in early summer and autumn, Agr. Forest Meteorol., 151, 682–691, https://doi.org/10.1016/j.agrformet.2010.12.010, 2011. a
Atkinson, R., Aschmann, S. M., Winer, A. M., and Carter, W. P. L.: Rate constants for the gas-phase reactions of nitrate radicals with furan, thiophene, and pyrrole at 295 ± 1 K and atmospheric pressure, Environ. Sci. Technol., 19, 87–90, https://doi.org/10.1021/es00131a010, 1985. a
Atkinson, R., Baulch, D. L., Cox, R. A., Hampson, R. F., Kerr, J. A., and Troe, J.: Evaluated Kinetic and Photochemical Data for Atmospheric Chemistry: Supplement IV, IUPAC Subcommittee on Gas Kinetic Data Evaluation for Atmospheric Chemistry, J. Phys. Chem. Ref. Data, 21, 1125–1568, https://doi.org/10.1063/1.555918, 1992. a
Bloss, C., Wagner, V., Jenkin, M. E., Volkamer, R., Bloss, W. J., Lee, J. D., Heard, D. E., Wirtz, K., Martin-Reviejo, M., Rea, G., Wenger, J. C., and Pilling, M. J.: Development of a detailed chemical mechanism (MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons, Atmos. Chem. Phys., 5, 641–664, https://doi.org/10.5194/acp-5-641-2005, 2005. a
Bourtsoukidis, E., Behrendt, T., Yañez-Serrano, A. M., Hellén, H., Diamantopoulos, E., Catão, E., Ashworth, K., Pozzer, A., Quesada, C. A., Martins, D. L., Sá, M., Araujo, A., Brito, J., Artaxo, P., Kesselmeier, J., Lelieveld, J., and Williams, J.: Strong sesquiterpene emissions from Amazonian soils, Nat. Commun., 9, 2226, https://doi.org/10.1038/s41467-018-04658-y, 2018. a
Boy, M., Rannik, U., Lehtinen, K. E. J., Tarvainen, V., Hakola, H., and Kulmala, M.: Nucleation events in the continental boundary layer: Long-term statistical analyses of aerosol relevant characteristics, J. Geophys. Res. Atmos., 108, 4667, https://doi.org/10.1029/2003JD003838, 2003. a
Boy, M., Sogachev, A., Lauros, J., Zhou, L., Guenther, A., and Smolander, S.: SOSA – a new model to simulate the concentrations of organic vapours and sulphuric acid inside the ABL – Part 1: Model description and initial evaluation, Atmos. Chem. Phys., 11, 43–51, https://doi.org/10.5194/acp-11-43-2011, 2011. a, b
Chen, X., Quéléver, L. L. J., Fung, P. L., Kesti, J., Rissanen, M. P., Bäck, J., Keronen, P., Junninen, H., Petäjä, T., Kerminen, V.-M., and Kulmala, M.: Observations of ozone depletion events in a Finnish boreal forest, Atmos. Chem. Phys., 18, 49–63, https://doi.org/10.5194/acp-18-49-2018, 2018. a, b
Damian, V., Sandu, A., Damian, M., Potra, F., and Carmichael, G. R.: The kinetic preprocessor KPP-a software environment for solving chemical kinetics, Comput. Chem. Eng., 26, 1567–1579, https://doi.org/10.1016/S0098-1354(02)00128-X, 2002. a
Di Carlo, P., Brune, W. H., Martinez, M., Harder, H., Lesher, R., Ren, X., Thornberry, T., Carroll, M. A., Young, V., Shepson, P. B., Riemer, D., Apel, E., and Campbell, C.: Missing OH Reactivity in a Forest: Evidence for Unknown Reactive Biogenic VOCs, Science, 304, 722–725, https://doi.org/10.1126/science.1094392, 2004. a
Donahue, N. M., Henry, K. M., Mentel, T. F., Kiendler-Scharr, A., Spindler, C., Bohn, B., Brauers, T., Dorn, H. P., Fuchs, H., Tillmann, R., Wahner, A., Saathoff, H., Naumann, K.-H., Möhler, O., Leisner, T., Müller, L., Reinnig, M.-C., Hoffmann, T., Salo, K., Hallquist, M., Frosch, M., Bilde, M., Tritscher, T., Barmet, P., Praplan, A. P., DeCarlo, P. F., Dommen, J., Prévôt, A. S. H., and Baltensperger, U.: Aging of biogenic secondary organic aerosol via gas-phase OH radical reactions, P. Natl. Acad. Sci. USA, 109, 13503–13508, https://doi.org/10.1073/pnas.1115186109, 2012. a
Edwards, P. M., Evans, M. J., Furneaux, K. L., Hopkins, J., Ingham, T., Jones, C., Lee, J. D., Lewis, A. C., Moller, S. J., Stone, D., Whalley, L. K., and Heard, D. E.: OH reactivity in a South East Asian tropical rainforest during the Oxidant and Particle Photochemical Processes (OP3) project, Atmos. Chem. Phys., 13, 9497–9514, https://doi.org/10.5194/acp-13-9497-2013, 2013. a, b
Ferracci, V., Heimann, I., Abraham, N. L., Pyle, J. A., and Archibald, A. T.: Global modelling of the total OH reactivity: investigations on the “missing” OH sink and its atmospheric implications, Atmos. Chem. Phys., 18, 7109–7129, https://doi.org/10.5194/acp-18-7109-2018, 2018. a
Fuchs, H., Novelli, A., Rolletter, M., Hofzumahaus, A., Pfannerstill, E. Y., Kessel, S., Edtbauer, A., Williams, J., Michoud, V., Dusanter, S., Locoge, N., Zannoni, N., Gros, V., Truong, F., Sarda-Esteve, R., Cryer, D. R., Brumby, C. A., Whalley, L. K., Stone, D., Seakins, P. W., Heard, D. E., Schoemaecker, C., Blocquet, M., Coudert, S., Batut, S., Fittschen, C., Thames, A. B., Brune, W. H., Ernest, C., Harder, H., Muller, J. B. A., Elste, T., Kubistin, D., Andres, S., Bohn, B., Hohaus, T., Holland, F., Li, X., Rohrer, F., Kiendler-Scharr, A., Tillmann, R., Wegener, R., Yu, Z., Zou, Q., and Wahner, A.: Comparison of OH reactivity measurements in the atmospheric simulation chamber SAPHIR, Atmos. Meas. Tech., 10, 4023–4053, https://doi.org/10.5194/amt-10-4023-2017, 2017. a
Guenther, A., Hewitt, C. N., Erickson, D., Fall, R., Geron, C., Graedel, T., Harley, P., Klinger, L., Lerdau, M., Mckay, W. A., Pierce, T., Scholes, B., Steinbrecher, R., Tallamraju, R., Taylor, J., and Zimmerman, P.: A global model of natural volatile organic compound emissions, J. Geophys. Res., 100, 8873–8892, https://doi.org/10.1029/94JD02950, 1995. a
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012. a
Hansen, R. F., Griffith, S. M., Dusanter, S., Rickly, P. S., Stevens, P. S., Bertman, S. B., Carroll, M. A., Erickson, M. H., Flynn, J. H., Grossberg, N., Jobson, B. T., Lefer, B. L., and Wallace, H. W.: Measurements of total hydroxyl radical reactivity during CABINEX 2009 – Part 1: field measurements, Atmos. Chem. Phys., 14, 2923-2937, https://doi.org/10.5194/acp-14-2923-2014, 2014. a
Hari, P. and Kulmala, M.: Station for Measuring Ecosystem-Atmosphere Relations (SMEAR II), Boreal Environ. Res., 10, 315–322, 2005. a, b
Hellén, H., Hakola, H., Pystynen, K.-H., Rinne, J., and Haapanala, S.: C2–C10 hydrocarbon emissions from a boreal wetland and forest floor, Biogeosciences, 3, 167–174, https://doi.org/10.5194/bg-3-167-2006, 2006. a
Hellén, H., Kuronen, P., and Hakola, H.: Heated stainless steel tube for ozone removal in the ambient air measurements of mono- and sesquiterpenes, Atmos. Environ., 57, 35–40, https://doi.org/10.1016/j.atmosenv.2012.04.019, 2012. a
Hellén, H., Schallhart, S., Praplan, A. P., Petäjä, T., and Hakola, H.: Using in situ GC-MS for analysis of C2–C7 volatile organic acids in ambient air of a boreal forest site, Atmos. Meas. Tech., 10, 281–289, https://doi.org/10.5194/amt-10-281-2017, 2017. a
Hellén, H., Praplan, A. P., Tykkä, T., Ylivinkka, I., Vakkari, V., Bäck, J., Petäjä, T., Kulmala, M., and Hakola, H.: Long-term measurements of volatile organic compounds highlight the importance of sesquiterpenes for the atmospheric chemistry of a boreal forest, Atmos. Chem. Phys., 18, 13839–13863, https://doi.org/10.5194/acp-18-13839-2018, 2018. a, b, c, d, e, f, g
Hemmilä, M., Hellén, H., Virkkula, A., Makkonen, U., Praplan, A. P., Kontkanen, J., Ahonen, L., Kulmala, M., and Hakola, H.: Amines in boreal forest air at SMEAR II station in Finland, Atmos. Chem. Phys., 18, 6367–6380, https://doi.org/10.5194/acp-18-6367-2018, 2018. a
Ilvesniemi, H., Levula, J., Ojansuu, R., Kolari, P., Kulmala, L., Pumpanen, J., Launiainen, S., Vesala, T., and Nikinmaa, E.: Long-term measurements of the carbon balance of a boreal Scots pine dominated forest ecosystem, Boreal Environ. Res., 14, 731–753, 2009. a
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997. a
Jenkin, M. E., Wyche, K. P., Evans, C. J., Carr, T., Monks, P. S., Alfarra, M. R., Barley, M. H., McFiggans, G. B., Young, J. C., and Rickard, A. R.: Development and chamber evaluation of the MCM v3.2 degradation scheme for β-caryophyllene, Atmos. Chem. Phys., 12, 5275–5308, https://doi.org/10.5194/acp-12-5275-2012, 2012. a
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015. a
Junninen, H., Lauri, A., Keronen, P., Aalto, P., Hiltunen, V., Hari, P., and Kulmala, M.: Smart-SMEAR: on-line data exploration and visualization tool for SMEAR stations, Boreal Env. Res., 14, 447–457, 2009. a
Kaiser, J., Skog, K. M., Baumann, K., Bertman, S. B., Brown, S. B., Brune, W. H., Crounse, J. D., de Gouw, J. A., Edgerton, E. S., Feiner, P. A., Goldstein, A. H., Koss, A., Misztal, P. K., Nguyen, T. B., Olson, K. F., St. Clair, J. M., Teng, A. P., Toma, S., Wennberg, P. O., Wild, R. J., Zhang, L., and Keutsch, F. N.: Speciation of OH reactivity above the canopy of an isoprene-dominated forest, Atmos. Chem. Phys., 16, 9349–9359, https://doi.org/10.5194/acp-16-9349-2016, 2016. a, b, c
Keenan, R. J., Reams, G. A., Achard, F., de Freitas, J. V., Grainger, A., and Lindquist, E.: Dynamics of global forest area: Results from the FAO Global Forest Resources Assessment 2015, For. Ecol. Manage., 352, 9–20, https://doi.org/10.1016/j.foreco.2015.06.014, 2015. a
Keßel, S.: Entwicklung und Charakterisierung der Comparative Reactivity Method zur Messung von Hydroxylradikal- und Chlorradikal-Reaktivitäten: troposphärische Oxidationschemie in drei unterschiedlich stark anthropogen beeinflussten Gebieten, 212 pp., 2016. a
Kim, S., Guenther, A., Karl, T., and Greenberg, J.: Contributions of primary and secondary biogenic VOC tototal OH reactivity during the CABINEX (Community Atmosphere-Biosphere INteractions Experiments)-09 field campaign, Atmos. Chem. Phys., 11, 8613–8623, https://doi.org/10.5194/acp-11-8613-2011, 2011. a, b
Kovacs, T. A. and Brune, W. H.: Total OH Loss Rate Measurement, J. Atmos. Chem., 39, 105–122, https://doi.org/10.1023/A:1010614113786, 2001. a
Kumar, V., Chandra, B. P., and Sinha, V.: Large unexplained suite of chemically reactive compounds present in ambient air due to biomass fires, Sci. Rep., 8, 626, https://doi.org/10.1038/s41598-017-19139-3, 2018. a, b
Leino, K., Riuttanen, L., Nieminen, T., Maso, M. D., Väänänen, R., Pohja, T., Keronen, P., Järvi, L., Aalto, P. P., Virkkula, A., Kerminen, V.-M., Petäjä, T., and Kulmala, M.: Biomass-burning smoke episodes in Finland from eastern European wildfires, Boreal Env. Res., 19, 275–292, 2014. a, b
Liebmann, J., Karu, E., Sobanski, N., Schuladen, J., Ehn, M., Schallhart, S., Quéléver, L., Hellen, H., Hakola, H., Hoffmann, T., Williams, J., Fischer, H., Lelieveld, J., and Crowley, J. N.: Direct measurement of NO3 radical reactivity in a boreal forest, Atmos. Chem. Phys., 18, 3799–3815, https://doi.org/10.5194/acp-18-3799-2018, 2018. a, b, c
Mao, J., Ren, X., Zhang, L., Van Duin, D. M., Cohen, R. C., Park, J.-H., Goldstein, A. H., Paulot, F., Beaver, M. R., Crounse, J. D., Wennberg, P. O., DiGangi, J. P., Henry, S. B., Keutsch, F. N., Park, C., Schade, G. W., Wolfe, G. M., Thornton, J. A., and Brune, W. H.: Insights into hydroxyl measurements and atmospheric oxidation in a California forest, Atmos. Chem. Phys., 12, 8009–8020, https://doi.org/10.5194/acp-12-8009-2012, 2012. a, b
Michoud, V., Hansen, R. F., Locoge, N., Stevens, P. S., and Dusanter, S.: Detailed characterizations of the new Mines Douai comparative reactivity method instrument via laboratory experiments and modeling, Atmos. Meas. Tech., 8, 3537–3553, https://doi.org/10.5194/amt-8-3537-2015, 2015. a, b, c
Mogensen, D., Smolander, S., Sogachev, A., Zhou, L., Sinha, V., Guenther, A., Williams, J., Nieminen, T., Kajos, M. K., Rinne, J., Kulmala, M., and Boy, M.: Modelling atmospheric OH-reactivity in a boreal forest ecosystem, Atmos. Chem. Phys., 11, 9709–9719, https://doi.org/10.5194/acp-11-9709-2011, 2011. a, b, c, d, e, f, g
Mogensen, D., Gierens, R., Crowley, J. N., Keronen, P., Smolander, S., Sogachev, A., Nölscher, A. C., Zhou, L., Kulmala, M., Tang, M. J., Williams, J., and Boy, M.: Simulations of atmospheric OH, O3 and NO3 reactivities within and above the boreal forest, Atmos. Chem. Phys., 15, 3909–3932, https://doi.org/10.5194/acp-15-3909-2015, 2015. a, b, c, d, e, f
Mäki, M., Heinonsalo, J., Hellén, H., and Bäck, J.: Contribution of understorey vegetation and soil processes to boreal forest isoprenoid exchange, Biogeosciences, 14, 1055–1073, https://doi.org/10.5194/bg-14-1055-2017, 2017. a
Mäki, M., Aaltonen, H., Heinonsalo, J., Hellén, H., Pumpanen, J., and Bäck, J.: Boreal forest soil is a significant and diverse source of volatile organic compounds, Plant Soil, 89–110, https://doi.org/10.1007/s11104-019-04092-z, 2019. a
Nakashima, Y., Kato, S., Greenberg, J., Harley, P., Karl, T., Turnipseed, A., Apel, E., Guenther, A., Smith, J., and Kajii, Y.: Total OH reactivity measurements in ambient air in a southern Rocky mountain ponderosa pine forest during BEACHON-SRM08 summer campaign, Atmos. Environ., 85, 1–8, https://doi.org/10.1016/j.atmosenv.2013.11.042, 2014. a, b
Nölscher, A. C., Williams, J., Sinha, V., Custer, T., Song, W., Johnson, A. M., Axinte, R., Bozem, H., Fischer, H., Pouvesle, N., Phillips, G., Crowley, J. N., Rantala, P., Rinne, J., Kulmala, M., Gonzales, D., Valverde-Canossa, J., Vogel, A., Hoffmann, T., Ouwersloot, H. G., Vilà-Guerau de Arellano, J., and Lelieveld, J.: Summertime total OH reactivity measurements from boreal forest during HUMPPA-COPEC 2010, Atmos. Chem. Phys., 12, 8257–8270, https://doi.org/10.5194/acp-12-8257-2012, 2012. a, b, c, d, e, f
Nölscher, A. C., Bourtsoukidis, E., Bonn, B., Kesselmeier, J., Lelieveld, J., and Williams, J.: Seasonal measurements of total OH reactivity emission rates from Norway spruce in 2011, Biogeosciences, 10, 4241–4257, https://doi.org/10.5194/bg-10-4241-2013, 2013. a
Nölscher, A. C., Yáñez-Serrano, A. M., Wolff, S., de Araujo, A. C., Lavrič, J. V., Kesselmeier, J., and Williams, J.: Unexpected seasonality in quantity and composition of Amazon rainforest air reactivity, Nat. Comm., 7, 10383, https://doi.org/10.1038/ncomms10383, 2016. a, b
Pearson, G., Davies, F., and Collier, C.: An Analysis of the Performance of the UFAM Pulsed Doppler Lidar for Observing the Boundary Layer, J. Atmos. Ocean. Tech., 26, 240–250, https://doi.org/10.1175/2008JTECHA1128.1, 2009. a
Praplan, A. P., Pfannerstill, E. Y., Williams, J., and Hellén, H.: OH reactivity of the urban air in Helsinki, Finland, during winter, Atmos. Environ., 169, 150–161, https://doi.org/10.1016/j.atmosenv.2017.09.013, 2017. a, b, c, d, e, f
Ramasamy, S., Ida, A., Jones, C., Kato, S., Tsurumaru, H., Kishimoto, I., Kawasaki, S., Sadanaga, Y., Nakashima, Y., Nakayama, T., Matsumi, Y., Mochida, M., Kagami, S., Deng, Y., Ogawa, S., Kawana, K., and Kajii, Y.: Total OH reactivity measurement in a BVOC dominated temperate forest during a summer campaign, 2014, Atmos. Environ., 131, 41–54, https://doi.org/10.1016/j.atmosenv.2016.01.039, 2016. a, b
Rossabi, S., Choudoir, M., Helmig, D., Hueber, J., and Fierer, N.: Volatile Organic Compound Emissions From Soil Following Wetting Events, J. Geophys. Res.-Biogeo., 123, 1988–2001, https://doi.org/10.1029/2018JG004514, 2018. a
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003. a
Sinha, V., Williams, J., Crowley, J. N., and Lelieveld, J.: The Comparative Reactivity Method – a new tool to measure total OH Reactivity in ambient air, Atmos. Chem. Phys., 8, 2213–2227, https://doi.org/10.5194/acp-8-2213-2008, 2008. a, b, c, d, e
Sinha, V., Williams, J., Lelieveld, J., Ruuskanen, T., Kajos, M., Patokoski, J., Hellen, H., Hakola, H., Mogensen, D., Boy, M., Rinne, J., and Kulmala, M.: OH Reactivity Measurements within a Boreal Forest: Evidence for Unknown Reactive Emissions, Environ. Sci. Technol., 44, 6614–6620, https://doi.org/10.1021/es101780b, 2010. a, b, c, d, e
Sogachev, A., Menzhulin, G. V., Heimann, M., and Lloyd, J.: A simple three-dimensional canopy – planetary boundary layer simulation model for scalar concentrations and fluxes, Tellus B, 54, 784–819, https://doi.org/10.3402/tellusb.v54i5.16729, 2002. a
Vakkari, V., O'Connor, E. J., Nisantzi, A., Mamouri, R. E., and Hadjimitsis, D. G.: Low-level mixing height detection in coastal locations with a scanning Doppler lidar, Atmos. Meas. Tech., 8, 1875–1885, https://doi.org/10.5194/amt-8-1875-2015, 2015. a
Williams, J. and Brune, W.: A roadmap for OH reactivity research, Atmos. Environ., 106, 371–372, https://doi.org/10.1016/j.atmosenv.2015.02.017, 2015. a
Yang, Y., Shao, M., Wang, X., Nölscher, A. C., Kessel, S., Guenther, A., and Williams, J.: Towards a quantitative understanding of total OH reactivity: A review, Atmos. Environ., 134, 147–161, https://doi.org/10.1016/j.atmosenv.2016.03.010, 2016. a
Zannoni, N., Dusanter, S., Gros, V., Sarda Esteve, R., Michoud, V., Sinha, V., Locoge, N., and Bonsang, B.: Intercomparison of two comparative reactivity method instruments inf the Mediterranean basin during summer 2013, Atmos. Meas. Tech., 8, 3851–3865, https://doi.org/10.5194/amt-8-3851-2015, 2015. a
Zannoni, N., Gros, V., Lanza, M., Sarda, R., Bonsang, B., Kalogridis, C., Preunkert, S., Legrand, M., Jambert, C., Boissard, C., and Lathiere, J.: OH reactivity and concentrations of biogenic volatile organic compounds in a Mediterranean forest of downy oak trees, Atmos. Chem. Phys., 16, 1619–1636, https://doi.org/10.5194/acp-16-1619-2016, 2016. a
Zhou, L., Nieminen, T., Mogensen, D., Smolander, S., Rusanen, A., Kulmala, M., and Boy, M.: SOSAA – A new model to simulate the concentrations of organic vapours, sulphuric acid and aerosols inside the ABL – Part 2: Aerosol dynamics and one case study at a boreal forest site, Boreal Env. Res., 19, 237–256, 2014. a
Zhou, P., Ganzeveld, L., Rannik, Ü., Zhou, L., Gierens, R., Taipale, D., Mammarella, I., and Boy, M.: Simulating ozone dry deposition at a boreal forest with a multi-layer canopy deposition model, Atmos. Chem. Phys., 17, 1361–1379, https://doi.org/10.5194/acp-17-1361-2017, 2017a. a
Zhou, P., Ganzeveld, L., Taipale, D., Rannik, Ü., Rantala, P., Rissanen, M. P., Chen, D., and Boy, M.: Boreal forest BVOC exchange: emissions versus in-canopy sinks, Atmos. Chem. Phys., 17, 14309–14332, https://doi.org/10.5194/acp-17-14309-2017, 2017b. a
- Abstract
- Introduction
- Methods
- Results and discussion
- Conclusions
- Data availability
- Appendix A: Pseudo 1st-order-kinetics correction
- Appendix B: Precision of the measurements
- Appendix C: Comparison of measured and modelled nopinone
- Appendix D: Details for the modelled periods
- Author contributions
- Competing interests
- Acknowledgements
- Financial support
- Review statement
- References
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
- Article
(16359 KB) - Full-text XML
- Abstract
- Introduction
- Methods
- Results and discussion
- Conclusions
- Data availability
- Appendix A: Pseudo 1st-order-kinetics correction
- Appendix B: Precision of the measurements
- Appendix C: Comparison of measured and modelled nopinone
- Appendix D: Details for the modelled periods
- Author contributions
- Competing interests
- Acknowledgements
- Financial support
- Review statement
- References