the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 04 Jan 2019
| 04 Jan 2019
Dynamic changes in optical and chemical properties of tar ball aerosols by atmospheric photochemical aging
Chunlin Li
Quanfu He
Julian Schade
Johannes Passig
Ralf Zimmermann
Daphne Meidan
Alexander Laskin
Following wood pyrolysis, tar ball aerosols were laboratory generated from wood tar separated into polar and nonpolar phases. Chemical information of fresh tar balls was obtained from a high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS) and single-particle laser desorption/resonance enhanced multiphoton ionization mass spectrometry (SP-LD-REMPI-MS). Their continuous refractive index (RI) between 365 and 425 nm was retrieved using a broadband cavity enhanced spectroscopy (BBCES). Dynamic changes in the optical and chemical properties for the nonpolar tar ball aerosols in NOx-dependent photochemical process were investigated in an oxidation flow reactor (OFR). Distinct differences in the chemical composition of the fresh polar and nonpolar tar aerosols were identified. Nonpolar tar aerosols contain predominantly high-molecular weight unsubstituted and alkyl-substituted polycylic aromatic hydrocarbons (PAHs), while polar tar aerosols consist of a high number of oxidized aromatic substances (e.g., methoxy-phenols, benzenediol) with higher O : C ratios and carbon oxidation states. Fresh tar balls have light absorption characteristics similar to atmospheric brown carbon (BrC) aerosol with higher absorption efficiency towards the UV wavelengths. The average retrieved RI is 1.661+0.020i and 1.635+0.003i for the nonpolar and polar tar aerosols, respectively, with an absorption Ångström exponent (AAE) between 5.7 and 7.8 in the detected wavelength range. The RI fits a volume mixing rule for internally mixed nonpolar/polar tar balls. The RI of the tar ball aerosols decreased with increasing wavelength under photochemical oxidation. Photolysis by UV light (254 nm), without strong oxidants in the system, slightly decreased the RI and increased the oxidation state of the tar balls. Oxidation under varying OH exposure levels and in the absence of NOx diminished the absorption (bleaching) and increased the O : C ratio of the tar balls. The photobleaching via OH radical initiated oxidation is mainly attributed to decomposition of chromophoric aromatics, nitrogen-containing organics, and high-molecular weight components in the aged particles. Photolysis of nitrous oxide (N2O) was used to simulate NOx-dependent photochemical aging of tar balls in the OFR. Under high-NOx conditions with similar OH exposure, photochemical aging led to the formation of organic nitrates, and increased both oxidation degree and light absorption for the aged tar ball aerosols. These observations suggest that secondary organic nitrate formation counteracts the bleaching by OH radical photooxidation to eventually regain some absorption of the aged tar ball aerosols. The atmospheric implication and climate effects from tar balls upon various oxidation processes are briefly discussed.
- Article
(7922 KB) -
Supplement
(6998 KB) - BibTeX
- EndNote





Organic aerosol (OA), which represent a ubiquitous and dominant burden of the tropospheric particulate pollutants, play important roles in atmospheric chemistry and balance of regional and global radiation (Jimenez et al., 2009; Kanakidou et al., 2005; Seinfeld and Pandis, 2016; Shrivastava et al., 2017). An indirect climate influence of OA relies on their interaction with water, thus acting as cloud condensation nuclei (CCN) that may alter the hydrological cycle (cloud formation and perception) and modify Earth's albedo (Forster and Taylor, 2006; IPCC, 2013; Seinfeld and Pandis, 2016). The direct climate effect of OA is through extinction of incoming solar radiation and outgoing longwave radiation. Of particular importance is the warming effect due to light-absorbing carbonaceous aerosol commonly termed brown carbon (BrC) (Andreae and Gelencsér, 2006). BrC is an important yet poorly understood OA component due to its complex physical properties, undefined chemical composition, and dynamic evolution under atmospheric processes (Adler et al., 2010; Moise et al., 2015; Laskin et al., 2015). It has been estimated that BrC accounts for 10 %–40 % of the total light absorption in the atmosphere and when deposited on snowpack (Bahadur et al., 2012; Park et al., 2010), and contributes a global forcing of 0.10–0.25 W m−2, with even higher values on regional scales (Feng et al., 2013).
The origin of BrC can be either primary (i.e., directly emitted) or secondary (i.e., generated by reactions of aromatic or carbonyl compounds in clouds or particles) (Laskin et al., 2015). On a global scale, biomass burning releases over two-thirds of primary BrC and also contributes substantially to overall secondary OA formation (Jacobson, 2014; Jo et al., 2016). Better understanding of the optical properties of biomass burning BrC aerosols is crucial for constraining its atmospheric and climatic implications and Earth's energy balance. Unlike black carbon that absorbs light strongly throughout the entire UV–visible range, different chromophores that may also be coupled via charge transfer complexes enable BrC absorption in a much more pronounced and complicated wavelength-dependence manner (Phillips and Smith, 2004; Reid et al., 2005; Lin et al., 2016, 2017).
Tar balls are a specific type of particle produced from wood combustion (especially from biomass smoldering burning) which is abundant in the troposphere (Pósfai et al., 2004; Hand et al., 2005; Chen et al., 2017). Tar ball particles have been collected and identified in many biomass burning plumes (Pósfai et al., 2004; Fu et al., 2012; Li et al., 2017). Microanalysis has found that tar balls are homogeneous spherical carbonaceous particles with sizes ranging from tens to hundreds of nanometers. These particles contribute a considerable fraction of the biomass burning BrC (Pósfai et al., 2004; Hand et al., 2005; Li et al., 2017). The estimated burden of tar balls on regional and global climatic forcing has been emphasized (Chung et al., 2012; Jacobson, 2012, 2014). Tar balls from different burning conditions and bio-fuels coexist with many other types of particles (e.g., inorganic salts, soot, and other carbonaceous aerosols in the form of internal or external mixing), and these smoke particles undergo rapid atmospheric processing once they are released from the fire (Pósfai et al., 2004; Hand et al., 2005; Li et al., 2015, 2017). However, in situ determination of the optical properties of these particles during their lifetimes in the air has seldom been reported due to inherent difficulty in selective tar ball sampling out of complex particle ensembles typical of field burning emissions.
The complex refractive index (; n and k are real and imaginary parts, corresponding to scattering and absorption, respectively) is an intrinsic optical property of aerosols. Quantifying the RI of OA is highly needed for evaluating the related radiative forcing influence (Moise et al., 2015). Recently, several studies have investigated the optical properties of tar ball particles (Chakrabarty et al., 2010; Hoffer et al., 2016; Sedlacek et al., 2018). The optical measurements reported for tar balls or other biomass burning BrC were discrete over several wavelengths that were constrained by instruments measuring particle light coefficients, or indirectly inferred from calculations based on their electron energy-loss spectra or from UV–visible absorption of solutions containing dissolved tar balls (Alexander et al., 2008). Hand et al. (2005) measured light scattering coefficients of tar ball-dominated fire plumes using a nephelometer, and reconstructed the scattering coefficients with simplified organic carbon (OC) and elemental carbon (EC) data to get an average RI of 1.56+0.02i for tar balls at λ=632 nm. Chakrabarty et al. (2010) measured the RI of tar ball particles from smoldering biomass combustion at 405, 532, and 780 nm; they observed a clear wavelength-, biofuel-, and even burning condition-dependent RI. The light absorption by tar balls was similar to humic-like substance (HULIS) with an imaginary part (0.002–0.015) that increased exponentially towards the near-UV wavelengths. Recently, Hoffer et al. (2016) generated tar ball particles from flameless wood pyrolysis in the laboratory. They reported a higher RI value of 1.84+0.20i at 550 nm, which fell closer to RI of soot than to that of HULIS. Large discrepancies reside in these results and discrete RI values make it difficult to decipher the complicated wavelength-dependence character of tar ball optical properties, which finally constrains the assessment of its radiative forcing effect.
Freshly emitted smoke BrC contains chromophores with diverse chemical structures, polarity, and volatility (Lin et al., 2016, 2017). After emission into the atmosphere, smoke particles undergo dynamic changes as a result of dilution, precipitation, and chemical processing on scales of seconds to days, which eventually affect the physiochemical properties of BrC particles during their lifetimes in the atmosphere (Reid et al., 2005; Li et al., 2015; Laskin et al., 2015). Sumlin et al. (2017) simulated atmospheric photooxidation of biomass burning BrC and reported that photooxidation diminishes their light absorption. Zhong and Jang (2014) investigated the influence of humidity and NOx presence in photochemical aging of biomass burning BrC. They found that sunlight faded the color of BrC and humidity facilitated the decay of light absorption by BrC, while the presence of NOx delayed the fading. Overall, they concluded that light absorption by BrC is governed by chromophore formation and bleaching by sunlight in the atmosphere. Therefore, evaluating the climatic impacts of tar ball particles requires more extensive investigation of its optical properties and understanding of the dynamic transformations of the optical properties during atmospheric aging.
In this study, we generated proxies for tar ball particles by flameless wood pyrolysis (Tóth et al., 2014; Hoffer et al., 2016). This method allows consistent and continuous generation of tar ball proxy aerosols for studying their properties and processes. The RI of the tar aerosols as a function of wavelength in the ultraviolet–short visible region (365–425 nm, 0.5 nm resolution) was determined using a broadband cavity enhanced spectrometer (BBCES). A high-resolution time of flight aerosol mass spectrometer (HR-ToF-AMS) and a single-particle mass spectrometer applying laser desorption/resonantly enhanced multiphoton ionization (SP-LD-REMPI-MS) were used for probing the chemical profile of tar ball aerosols under NOx-dependent multiple-day photochemical oxidation. Specifically, the dynamic changes in their optical properties in correlation with their chemical composition were investigated. The atmospheric implications and climate forcing due to atmospheric aging of tar aerosols and evolution of their optical properties were also explored.
2.1 Tar ball particle generation
Following the formation mechanism in biomass burning process, polydisperse tar ball particles were generated from droplets of wood tar in the laboratory (Tóth et al., 2014; Hoffer et al., 2016). In this study, a similar procedure was applied for producing tar ball aerosols. In brief, commercial wood pellets (Hallingdal Trepellets, water content of 6.55 wt %, size of 2–3 cm in length, 0.2–0.3 cm in diameter) were smashed, heated and dry-distilled in the absence of air (25 ∘C min−1 increase to 530 ∘C from room temperature, and held for 20 min at 530 ∘C) to produce liquid tar-water emulsions (∼25 mL per hundred grams of used wood pellets). The emulsions were filtrated using 0.45 µm pore size filters (PTFE membrane, diameter 47 mm, Pall Corp.) to remove particulate matter or solid precipitation. After overnight static stabilization, the wood tar solution was phase-separated into water soluble and non-soluble oily phases at an initial 3 : 1 volume ratio. Herein, we will term these two fractions as “polar” and “nonpolar” phases, respectively. The phase-separated solution was further concentrated using a heating plate at 300 ∘C with N2 purge flow to prevent oxidation. A final 1 : 1 volume ratio of polar to nonpolar phase was obtained; then the concentrated solutions were sealed and stored in the dark under 2 ∘C for following experiments. With respect to their potential reactivity and instability, the distillation products were used within a few days.
For particle optical measurement, tar balls were produced from aerosolization of above predefined wood tar diluted in methanol (Gradient grade for HPLC, purity ≥ 99.9 wt %, Merck) using a constant output atomizer (Model 3076, TSI) with high-purity N2 as a carrier gas. As the actual fractions of the polar and nonpolar compounds contributing to the mass of ambient tar ball or biomass burning organic aerosol (BBOA) can vary with biofuel sources, burning condition, atmospheric process, and also method/efficiency to classify the polar and nonpolar materials from the sample (Sengupta et al., 2018; Lin et al., 2017, 2018; Chen and Bond, 2010; Rajput et al., 2014), tar ball aerosols in this study were generated from polar, nonpolar, and mixtures of these two phase tar solutions at volume mixing ratio of 2:1, 1:1, and 1:2, respectively. Activated charcoal denuders and quartz heating tube (150 ∘C, residence time ∼0.7s for particles at a nitrogen flow of 1.0 L min−1) were used after the atomizer to outgas the methanol from the gaseous and particulate phases. Mesh filters (TSI) downstream were used to filter out some ultrafine (less than 100 nm) particles.
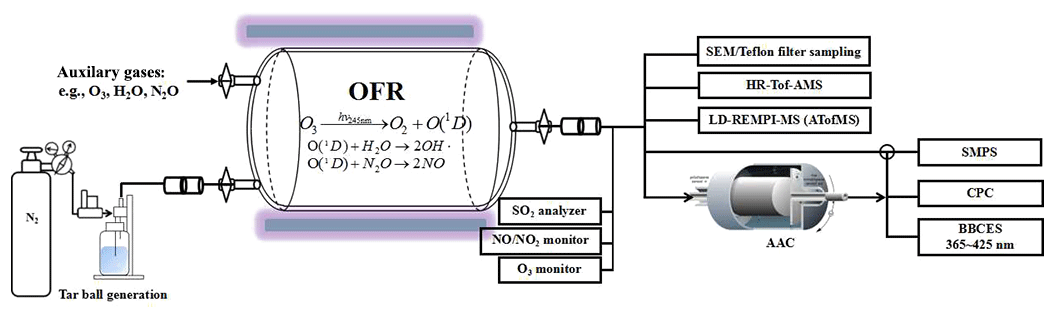
Figure 1Experimental setup for laboratory generation and aging of tar ball aerosol, including generation setup, OFR photochemical aging, gaseous–particulate chemical monitoring, particle size distribution and optical property measurements.
2.2 NOx-dependent OH oxidation of tar ball aerosols
Heterogeneous oxidation of tar ball aerosols was simulated using an oxidation flow reactor (OFR), shown schematically in Fig. 1. The OFR has been characterized (Kang et al., 2007; Peng et al., 2015, 2016) and the operational procedures have been described previously (He et al., 2018). Briefly, the OFR consists of a horizontal 13.3 L aluminum cylindrical chamber (46 cm long × 22 cm ID) operating in continuous flow mode. The chamber is equipped with two power-controllable ozone-free mercury lamps with peak emission at λ=254 nm (82-934-08, BHK Inc., CA, USA). The two UV lamps are surrounded by Pyrex sheath tubes that are continuously purged with N2 to cool the lamps and remove outgassing compounds. OH radicals in the OFR are produced through photolysis of externally introduced O3 under 254 nm illumination and further reaction of singlet oxygen (O1D) with water vapor:
External O3 was produced by irradiation of 0.2 L min−1 high-purity O2 using a mercury lamp (λ=185 nm, 78-2046-07, BHK Inc., CA). The O3 concentration downstream of the OFR was measured by an O3 monitor (2B Technology). A Nafion membrane humidifier (Perma Pure LIC, NJ) was used to supply water vapor to the OFR. Tar ball aerosols carried by 1.0 L min−1 N2 flow from the atomizer were introduced into OFR. The initial aerosol concentrations in the OFR were mediated by controlling the concentration of the wood tar solution to be atomized until the number of 350 nm particles was above 100 cm−3, as shown in Fig. S1 in the Supplement of tar ball aerosol size distribution. Finally, a total flow of 5.5 L min−1 with 36 %–38 % RH, initial 27–28 ppm O3, and 200–250 µg m−3 tar ball particles (assuming material density of 1.0 g cm−3) was maintained, with a corresponding plug flow residence time (RT) of 144 s for aerosols in the OFR.
The extent of simulated daytime oxidation by OH exposure was varied by changing the UV light intensity. Here, OH exposures in the OFR were inferred by measuring the decay of added SO2 (monitored by Thermo SO2 analyzer, model 43i) due to reaction with OH radicals at specific UV lamp intensity. A low concentration (∼60 ppb) of SO2 was used to minimize its influence on the OH radical reactivity. Typical total OH exposures ranged from to molec cm−3 s or 0.5–7 equivalent daytime atmospheric oxidation days (EAD) were maintained, taking typical ambient average OH concentration as 1.5×106 molec cm−3 (Kang et al., 2007; Peng et al., 2015, 2016).
In addition to reactions with oxidants, organic aerosols may change their chemical and physical properties by photolysis (Epstein et al., 2014; Lee et al., 2014; Wong et al., 2014). Therefore, the influence of light irradiation during tar ball photochemical aging was assessed at the short exposure time in the OFR. Here, tar ball aging was repeated at the same conditions (e.g., RT, RH, N2 / O2 flow, tar ball concentration, UV lamp power) without O3 supply in the OFR. The 254 nm photon flux at specific to maximal UV lamp power was calculated by fitting the OH exposure estimated from SO2 decay and by the Aerodyne OFR Exposure Estimator (v3.1, https://sites.google.com/site/pamwiki/hardware/estimation-equations, last access: 20 December 2017).
Under polluted conditions, nitrogen oxides (NOx) are often involved in the atmospheric transformations of organic aerosol and alter their physiochemical properties (Rollins et al., 2012; Ng et al., 2007; Lin et al., 2015). Therefore, NOx influence on tar ball aerosol aging was also investigated. Due to rapid conversion of NOx (NO + NO2) into nitric acid (HNO3) under high O3 and OH concentrations, simple addition of NOx into OFR cannot sustain NOx levels that compete with HO2 radicals in the reaction with organic proxy (ROO). NOx generated via N2O reaction with O1D has been modeled and tested to suit the characterization of NOx-dependent SOA formation pathways using OFR (Peng et al., 2017; Lambe et al., 2017). In this study, N2O (99.999 %) addition of 0.5 % vol and 2.0 % vol were used during tar ball aerosol photochemical oxidation in the OFR, and equivalent OH exposure of about 4.0 EAD was maintained. NOx (NO and NO2) concentrations downstream of the OFR was measured using a NO / NO2 analyzer (Ecotech, Serinus 40 NOx). Experimental parameters including initial O3 and N2O concentrations, NOx, moisture ratio, maintained OH exposures and the corresponding photon flux at 254 nm are presented in Table 1.
Table 1Experimental conditions for tar ball particles photochemical oxidation.
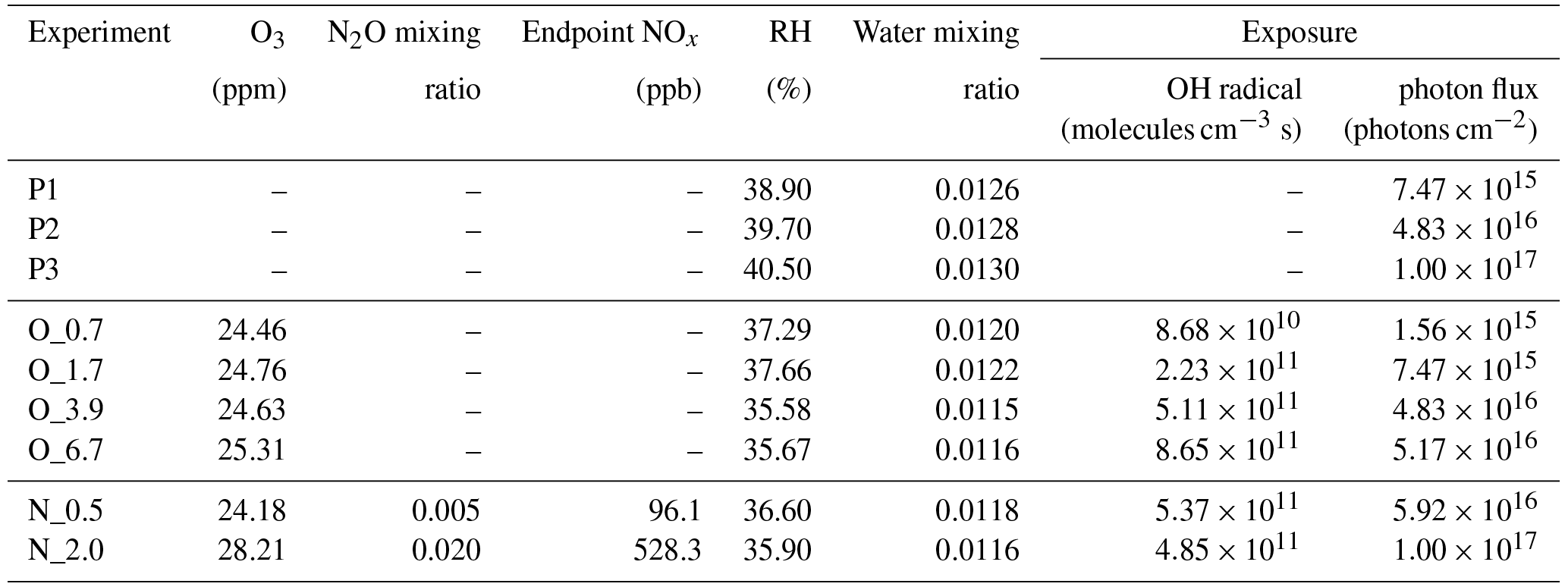
Note: P1–P3 mean photolysis test, O_0.7–O_6.7 correspond to the photochemical oxidation experiment from equivalent 0.7 to 6.7 days' ageing, and N_0.5 and N_2.0 indicate photochemical oxidation with N2O addition at 0.5 % vol and 2 % vol mixing ratios (standard deviation for the parameters was not given in above table).
2.3 Online optical and chemical characterization
Prior to the optical and chemical measurements, excess ozone and NOx were removed from the sample air stream following the OFR using two diffusion denuders packed with Carulite (Carus Corporation, Peru, IL) and one activated charcoal tube. The streamflow was further dehydrated with two silica gel diffusion dryers. Afterward, the tar ball aerosols were characterized by a combination of online chemical and optical instruments.
Bulk chemical fragments and organic elemental ratios of tar ball aerosols were monitored in real time by the HR-ToF-AMS (Aerodyne Research Inc., Billerica, MA, USA) in alternating high-sensitivity V and high-resolution W modes. The working principles of the AMS have been described in detail elsewhere (DeCarlo et al., 2006). In short, aerosol particles are separated from the gas phase through an aerodynamic lens system and then transferred into the vacuum system, where they are impacted onto a vaporizer at about 600 ∘C, thus vaporizing the particles. The analyte vapors are ionized with 70 eV electron impact ionization (EI). A time-of-flight mass spectrometer is used for high-resolution analysis of the ions. SQUIRREL v1.16 and PIKA v1.57 codes (http://cires.colorado.edu/jimenez-group/ToFAMSResources/ToFSoftware/, last access: 24 November 2016) were used to process the collected AMS data. Four ion groups were classified as Cx, CxHyO+, CxHy (z > 1), and CxHyOi(i0, p1) based on fragment features. The ions O+, OH+, and H2O+ were included in the CxHy group, as concentrations of these species were calculated from the organic ion abundance using the method in Aiken et al. (2008). The ambient improved (AI) atomic ratios of oxygen to carbon (O : C), hydrogen to carbon (H : C), nitrogen to carbon (N : C), and organic mass to organic carbon (OM ∕ OC) were generated from the measured ion fragments.
Particle-bound organic molecules were measured using a custom single-particle time-of-flight mass spectrometer. This instrument features laser desorption and resonantly enhanced multiphoton ionization (SP-LD-REMPI-ToF-MS), allowing for the detection of aromatic substances on individual particles. Detailed description and application of the instrument in LD-REMPI ionization mode is given by Bente et al. (2008) and Passig et al. (2017). Briefly, aerodynamically accelerated particles are individually sized using laser velocimetry, and heated by a pulsed CO2 infrared laser (10.6 µm) to desorb organic molecules. Aromatic substances in the gas plume are selectively ionized via REMPI by a KrF-excimer laser pulse (248 nm) and detected in the positive MS flight tube. The REMPI-MS technique is very sensitive and selective for aromatic substances (Boesl et al., 1978; Grotemeyer et al., 1986; Rettner and Brophy, 1981) and suitable for studies on pyrolysis and (wood) combustion processes (Heger et al., 1999; Czech et al., 2017). For the tar ball aerosols it provides complementary information to the HR-ToF-AMS spectra. A custom software on LabView basis records and calculates the aerodynamic size and individual mass spectra of the particles.
For optical measurements, tar ball aerosols were size-selected using an Aerosol Aerodynamic Classifier (AAC, Cambustion, UK). AAC has significant advantages over the commonly used Differential Mobility Analyzer (DMA) classifier. The AAC classifies particles based on the aerodynamic size without charging and hence it avoids the contribution of multiply charged particles, thus generating real monodisperse size-selected particles distribution, reducing the errors associated with multiply charged large particles. In addition, the AAC has higher particle transmission efficiency at the relevant size range (Tavakoli and Olfert, 2013, 2014). Aerodynamic size-classified particles after the AAC were further scanned by a scanning mobility particle sizer (SMPS, classifier Model 3080, DMA Model 3081, CPC model 3775, TSI) to derive their mobility size distribution. The effective density of tar balls can be estimated from Eq. (1) with assumptions of homogeneous composition and particle shape factor of 1.0, which was verified later in this study:
where ρeff is an effective density, and Daero and Dm are aerodynamic and mobility diameters, respectively. ρo is unit density of 1.0 g cm−3.
Based on the derived effective density, size-specific tar ball aerosols covering mobility diameters between 175 to 350 nm with an interval of 25 nm were size-selected via AAC, and monodisperse tar balls were introduced into a dual-channel broad-band cavity enhanced spectrometer (BBCES) for light extinction (αext) measurements in the wavelength of 360–395 and 385–435 nm (at resolution 0.5 nm). A detailed description of the instrument can be found elsewhere (Washenfelder et al., 2013; Flores et al., 2014a, b). With the combination of a condensation particle counter (CPC, Model 3575, TSI) to measure particle concentration (N) in series, size-specific particle extinction cross section (σext) can be calculated by Eq. (2):
where λ is the wavelength of incidence light and Dp is the particle mobility diameter.
Using the Mie–Lorenz scattering theory, the wavelength-dependent complex refractive index of spherical homogeneous particles was derived (Pettersson et al., 2004; Abo Riziq et al., 2007). The retrieval algorithm was limited to search for n1 and k0 as their physical boundaries. Thereafter, spectral dependent extinction, scattering, and absorption cross sections (σext, σsca, and σabs) were calculated from the complex RI at a specific particle size. Using these parameters, the single scattering albedo, indicating the scattering fraction of light extinction (SSA , was calculated.
The absorption and extinction Ångström exponents (Åabs and Åext) describe the spectral dependence of aerosol light properties, and are widely used in climate modeling (Russell et al., 2010). It is customary to extrapolate the optical spectral absorption and extinction fitting to the range of wavelengths using a power law and , respectively. In this work, we determined Åext and Åabs with a linear regression of ln(σext) and ln(σabs) against ln(λ) over the range of 365 to 425 nm:
Here Åext, Åabs, and SSA were calculated for tar ball aerosols with a median diameter of 150 nm.
2.4 Offline optical characterization
In addition to the in situ measurements, tar ball particles were also collected quantitatively onto Teflon filters (47 mm diameter, 0.45 µm porosity, Pall Corp.) at a sampling flow rate of 2 L min−1 and then extracted using methanol (HPLC grade, purity ≥99.9 %, Merck) for offline UV–visible absorption measurement (Cary 60 UV-VIS spectroscopy, Agilent). Methanol extraction of organic compounds has been commonly performed in various studies (Hoffer et al., 2006; Laskin et al., 2009; Yee et al., 2013; Finewax et al., 2018; Xie et al., 2017). Here we verified the completeness of the extraction by extracting each filter twice with methanol. Moreover, vortex shaking (Vortex Genie-2, Scientific Industries) rather than sonication was applied to avoid chemical degradation of the extracts upon ultrasonic irradiation (Miljevic et al., 2014; Mutzel et al., 2013). The methanol extractable BrC mass absorption cross section (MAC, m2 g−1) and refractive imaginary k of the tar balls were estimated based on the following relations (Chen and Bond, 2010; Laskin et al., 2015):
Abs(λ) is the base-10 absorbance result from UV–visible spectroscopy (unitless), b is the optical length of the solution (1 cm), and C is the extracted organic carbon mass concentration in solvent (g m−3), which can be determined directly by normalizing the extract concentration and OC mass fraction for tar balls as OC ∕ OM obtained from AMS data, as no other refractory elemental carbon (EC) content was detected in our samples (see details in the Supplement). λ is the incident light wavelength, and ρ is the material density (g cm−3). Here, the derived effective density ρeff was used. The absorption Ångström exponent based on MAC was also derived as Å over the 365–425 nm spectral range.
In addition, particles were impacted at a flow of 2.5 L min−1 onto cyclopore track-etched polycarbonate membrane (47 mm, 0.1 µm porosity, Whatman Inc.) to investigate the morphology of tar balls using scanning electronic microscopy (SEM, JEOL JSM-7000F).
2.5 Radiative impacts of tar ball aerosols
To assess the climatic influence of tar ball aerosols, a wavelength-dependent direct shortwave aerosol simple radiative forcing efficiency (SRF, W g−1) was estimated using the clear sky air mass global horizonal solar spectrum (AM1GH), assuming that tar ball aerosols form a uniform, optically thin aerosol layer at the lower troposphere or on ground (Bond and Bergstrom, 2006; Levinson et al., 2010):
where dS(λ)∕dλ is the solar irradiance (photons s−1 cm−2), τatm is the atmospheric transmission (taking 0.79 for simple calculation), Fc is the cloud fraction (approximately 0.6), Rsfc is the surface albedo (approximately 0.19 for urban area ground and 0.8 for snow) (Chen and Bond, 2010), β is the average up-scatter fraction (the fraction of scattered sunlight that is scattered into the upward hemisphere), and MSC(λ) is the wavelength-dependent mass scattering cross section, respectively. We simply calculated radiative forcing of particles with an atmospheric-relevant size of 50 to 500 nm, and SRF was estimated and integrated over the measured range of 365–425 nm. The actinic flux over 365–425 nm was obtained from the “Quick TUV Calculator”, available at http://cprm.acom.ucar.edu/Models/TUV/Interactive_TUV/ (last access: 15 May 2018) using the following parameters: SZA (solar zenith angle) of 0∘, noon time, 30 June 2000, 300 Dobson overhead ozone column, surface albedo of 0.19 for urban area and 0.8 for snow, and 0 km altitude.
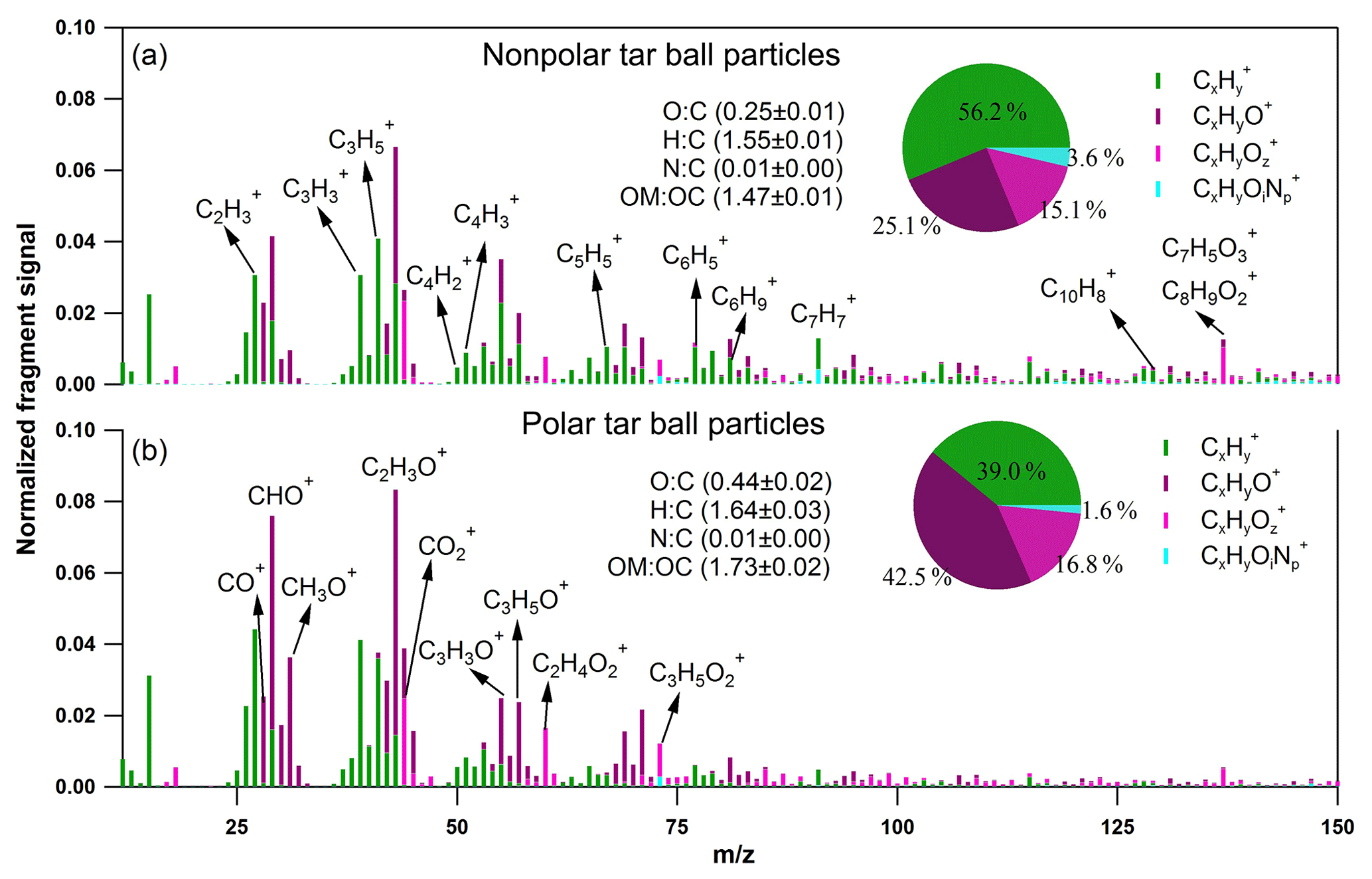
Figure 2High-resolution AMS mass spectra of fresh polar and nonpolar tar ball particles. Four ion groups are grouped for clarity as Cx (green), CxHyO+ (purple), CxHy (z > 1) (violet), and CxHyOi (i0, p1) (light blue). The mass fractions of the four fragment groups are presented by pie charts.
3.1 Chemical composition and optical properties of fresh tar ball aerosols
Negligible fractions of inorganics (e.g., sulfate, nitrate, chloride, and ammonium) in tar balls are obtained from AMS measurement as shown in Fig. S2, and these results confirm again that tar ball aerosols contain dominated carbonaceous compounds with minor amounts of N, S, and Cl (Pósfai et al., 2004; Hand et al., 2005; Adachi and Buseck, 2011). Thereafter, only organics in tar balls are considered, and the high-resolution bulk organic mass spectra for polar and nonpolar tar ball particles are given in Fig. 2. The mass spectra features and particle effective densities are summarized and compared in Table S1 in the Supplement. Distinct differences in the chemical composition were observed between polar and nonpolar tar ball aerosols. The alkyl fragments (Cx, e.g., Cn, Cn) dominate the signals for nonpolar particles (accounting for ∼56 % of the total fragments), implying that the nonpolar tar balls have compositional similarity to common hydrocarbon organic aerosol (HOA). The CxHyO+ fragments are the primary ions for the polar tar balls, contributing ∼42 % of their mass spectrum, suggesting that most of the organic constituents in the polar tar balls are substantially oxygenated. Both spectra exhibit significant intensity at m∕z 28 (CO+), m∕z 29 (CHO+), and m∕z 43 (C2H3O+), indicating the presence of carbonyl ions. The strong signal at m∕z 31 (CH3O+) results from methoxy species that preferably partition into the polar tar fraction. In addition, the significant signals at m∕z 50–52 (C4, C4, C4), m∕z 65 (C5), m∕z 77–78 (, C6), m∕z 81 (C6), and m∕z 91 (C7), which are characteristic of aromatic compounds, indicate that tar balls, especially from the nonpolar phase, contain a considerable amount of aromatic organics or present high aromaticity. Ion peaks at m∕z 77–78, 81, and 91 are typical of monocyclic aromatics such as alkyl-substituted benzene (for m∕z 77–78, 91) and heterocyclic aromatics (for m∕z 81) (Li et al., 2012), while the relatively higher signal at m∕z 128 (C10) in the nonpolar tar ball mass spectra can be assigned to the molecular ion of naphthalene (Herring et al., 2015). Moreover, signals at m∕z 55 and 57 (C3H3O+, C3H5O+) are signature fragments of aliphatic and non-acid oxygenated organics that are used to trace cooking emissions (He et al., 2010), and these two fragments were also observed in the tar ball aerosols. Similar to ambient biomass burning emissions, C2H4 (m∕z 60) and C3H5 (m∕z 73), two characteristic fragments from levoglucosan and similar cellulose pyrolyzed species (e.g., mannosan, galactosan) were detected in all the tar ball aerosols, and these fragments were more prominent in the polar aerosols due to the solubility of levoglucosan and analogs in water. Weimer et al. (2008) reported the percentage of m∕z 60 and 73 for the burning of various woods to be 0.6 %–4.1 % and 0.1 %–2.0 %, respectively. The percentages of these two fragments in our tar ball aerosols (0.7 %–1.6 % for m∕z 60 and 0.5 %–0.9 % for m∕z 73) are comparable to the literature data, although the fuel and the pyrolysis procedure are different. The m∕z 137 peak is dominated by fragments of C8H9 and C7H5, and these fragments have been determined in biomass burning emissions and were assigned to lignin-related ions with methoxy-phenolic structures (Li et al., 2012, 2014). Phenols and methoxy phenols are prominent compounds, accounting for 41 % of the identified organic species in primary BBOA (Schauer et al., 2001). The signal at m∕z 137 is much higher in the nonpolar-phase tar ball aerosols (1.0 % and 0.5 % for nonpolar and polar tar balls, respectively), and the fraction of fragment m∕z 137 is consistent with reference values of 0.3 %–2.0 % (Li et al., 2012). m∕z 44 (), a marker fragment of carboxylic acids, has been parameterized as f44 (fraction of a mass spectrum signal at m∕z 44) to present the oxidation degree of organic aerosols (Aiken et al., 2008; Ng et al., 2010). Higher f44 values indicate more oxidized OA (OOA), while less oxidized OA is characterized by lower f44 (Schauer et al., 2001). Peroxides can also produce a signal via extensive fragmentation in the AMS (Aiken et al., 2008). f44 has also been shown to be linearly correlated with the elemental O : C ratio of OA (Aiken et al., 2008). In this study, f44 for the nonpolar and polar tar ball aerosols are 1.9 % and 2.4 %, and the corresponding O : C ratios are 0.25 and 0.44. The higher O : C and H : C ratios explain the polarity of the polar tar ball aerosols. The simplified average carbon oxidation state (O : C–H : C) describes the oxidation level of particulate complex organic mixtures (Kroll et al., 2011). The calculated for the tar ball aerosols are −1.05–0.76, which agrees well with reference values of −1.0–0.7 for primary BBOA (Kroll et al., 2011). These values are in the broad range of −1.7–1.6 for HOA and −0.5–0 for semivolatile OOA (Aiken et al., 2008). In addition, a small fraction of nitrogen-containing organic compounds (NOCs) was detected, with the CxHyOi group contributing 1.6 %–3.6 % of the tar ball mass spectra with an estimated N : C ratio below 0.01, which agrees with previously reported N : C values of 0.008–0.018 for biomass burning emissions (He et al., 2010). Biomass burning is an important source of NOCs in the atmosphere, and alkaloid and nitro-aromatic constituents were detected to be abundant constituents of the NOCs (Laskin et al., 2009; Lin et al., 2017). Nitroaromatic compounds were also identified in urban fire emissions (Bluvshtein et al., 2017; Lin et al., 2017). Although these compounds constitute a small fraction of the BBOA mass, these chromophoric NOC species accounted for 50 %–80 % of the total visible light absorption by the extractable BrC (Lin et al., 2017).
Fragments larger than 100 amu () contribute a large fraction of the total organic signals for tar ball aerosols, consistent with biomass burning emissions that contain a large fraction of high-molecular weight compounds (Ge et al., 2012; Zhou et al., 2017). is 32 % for the nonpolar tar ball aerosols, which is higher than that of the polar particles (15 %), demonstrating that the nonpolar tar balls consist of more high-molecular weight organics. The measured effective densities for polar and nonpolar tar balls are 1.33 and 1.24 g cm−3. Chemical characteristics from AMS and densities for internal mixture tar balls follow the volume-linear mixing of polar and nonpolar tar solutions.
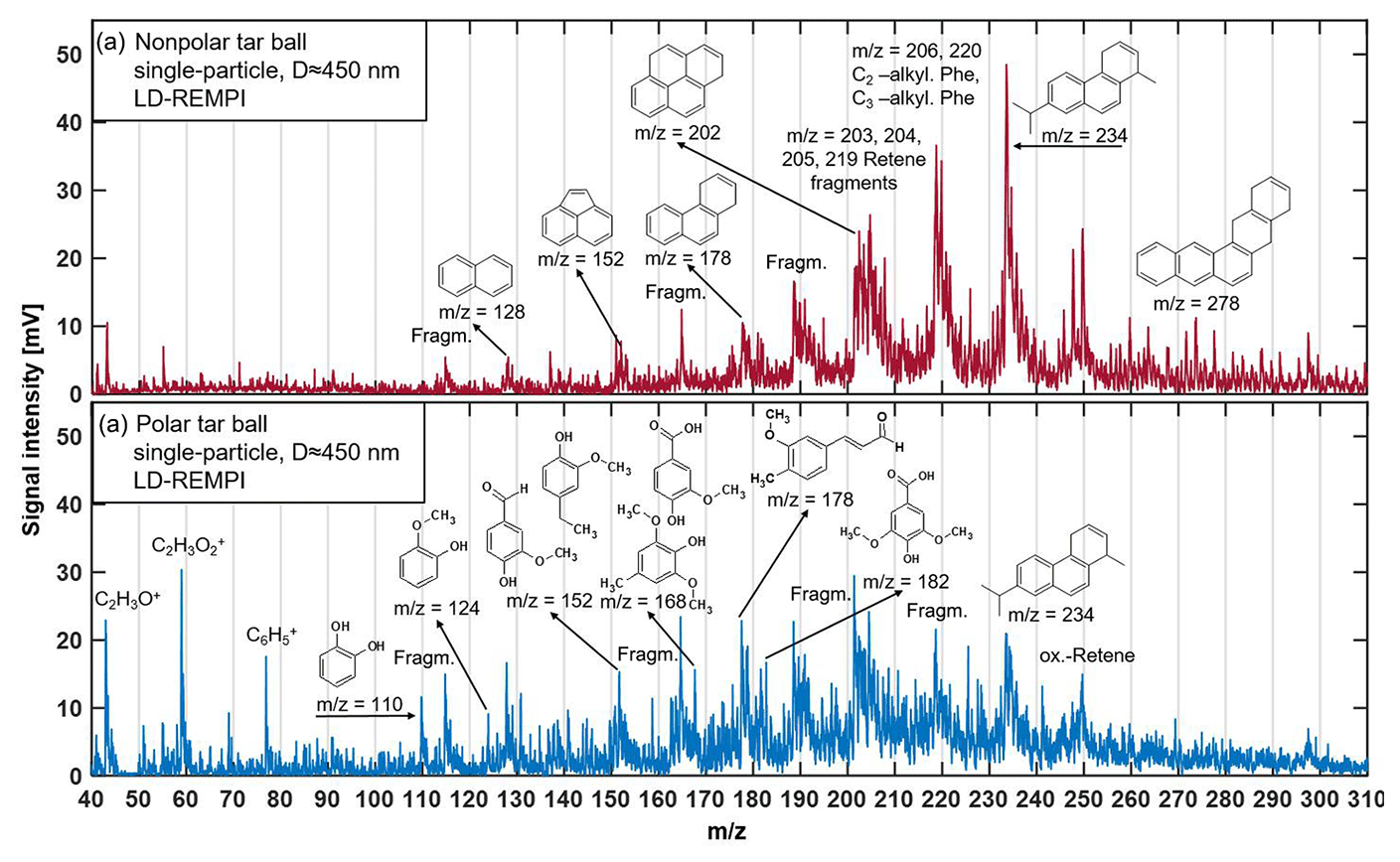
Figure 3LD-REMPI mass spectra of exemplary single tar ball particles; some feature peaks were identified and labeled. (a) Nonpolar tar ball spectra show predominantly alkyl-substituted and unsubstituted PAHs. (b) Polar tar ball spectra reveal many oxidized aromatics, e.g., methoxy-phenol and benzenediol. Note the softwood combustion marker retene at 234, its characteristic fragments ( 203, 204, 205, 219), and possible retene derivatives ( 248, 250).
A considerable aromatic fraction in the tar ball aerosols was confirmed by the LD-REMPI-MS measurement. Fig. 3 presents the mass spectra of aromatic substances obtained for one exemplary polar and one nonpolar particle, respectively. Aerodynamic size distributions for the detected tar ball aerosols are given in Fig. S3, and substances identified in the mass spectra are listed in Table S2. The features in the mass spectra are consistent with the polarity of examined tar ball aerosols and ambient BBOA (Lin et al., 2018). The complex REMPI spectrum shows rows of intense unsubstituted and partially alkylated PAH peaks in the nonpolar tar balls, including naphthalene, acenaphthylene, phenanthrene, pyrene, and, in particular, the softwood combustion marker retene at m∕z 234 and some possible derivatives (oxidized retene at m∕z 250 with one oxygen addition, methyl retene at m∕z 248 with one methyl addition) (McDonald et al., 2000; Shen et al., 2012). Retene and some of the aforementioned PAHs are also observed in the polar tar ball aerosols' mass spectra with lower intensities. In contrast, the polar tar ball REMPI mass spectra show strong peaks from oxidized aromatics, more specifically, benzenediol and methoxy-phenols (e.g., catechol, guaiacol, acetovanillone, syringaldehyde, conifery aldehyde). These results are also verified in ambient BBOA, of which the nonpolar BrC consists of primary unsubstituted PAHs, while the polar fraction includes major aromatic acids and phenols (Lin et al., 2018). The REMPI mass spectra correspond to the large fractions of CHO+ and CH3O+ fragments and high O : C ratios observed for the polar tar aerosols via HR-ToF-AMS, and remain consistent with the strong signals of typical aromatic fragments observed in the nonpolar tar aerosols in Fig. 2. The dominance of aromatic compounds in tar ball chemical composition agrees well with previous work on BBOA (Schauer et al., 2001; Wei et al., 2015; Bente et al., 2008, 2009; Czech et al., 2017). Biomass burning is a major source of environmental PAHs (alkylated PAHs, oxygenated PAHs, phenols, nitrogen-substituted PAHs, etc.) in both particulate and gaseous phases, and extensive emissions of PAHs from incomplete combustion pose a great threat to ecosystem and human health due to their carcinogenic toxicity (Li et al., 2017; Shen et al., 2013; Sigsgaard et al., 2015; Shrivastava et al., 2017). Moreover, the primary PAHs can act as precursors that substantially contribute to ambient SOA or BrC aerosol when involved in atmospheric photochemical aging, leading to profound climatic influence (Yee et al., 2013; Yu et al., 2014; Lu et al., 2011; Zhang et al., 2012).

Figure 4Wavelength-dependent RI and SSA for tar ball particles generated from polar, nonpolar and a mixture of the two-phase tarry solutions (only retrieval for a mixture of 1 : 1 in vol is shown for clarity; optical results for the other two mixtures can be found in the supporting materials). The shaded areas indicate the upper and lower limits of the imaginary part calculated from UV–visible spectra of methanol extracts from the corresponding tar ball particle samples: (a) real part, (b) imaginary part, and (c) SSA calculated for 150 nm particles. Overlaid in green symbols are previous measurements of biomass burning from the literature.
The complex refractive index (RI) of tar ball aerosols was retrieved under the assumption that the particles have similar chemical composition and a spherical shape. The SEM images shown in Fig. S4 confirm the spherical morphology and homogeneous composition of the tar ball particles generated in this study. Electron energy-loss spectroscopy (EELS) spectra indicates that the tar ball particles contain major C and minor O, which fits the AMS result and previous work (Pósfai et al., 2004; Chakrabarty et al., 2010). Continuous spectral-dependent RI and SSA for tar balls were derived and are presented in Fig. 4, RI results for tar ball aerosol at mixing ratio of 2 : 1 and 1 : 2 are presented in Fig. S5. Although scattering dominates the light extinction, absorption in the UV and in the visible ranges was unambiguously identified for the tar ball aerosols, with characteristic absorption similar to atmospheric BrC and HULIS (Hoffer et al., 2006; Bluvshtein et al., 2017; Lin et al., 2017). The imaginary part, k, increases towards the UV range, presenting 0.02–0.03 difference over the measured spectra range. The real part, n, for the nonpolar tar balls decreased from 1.673 at 365 nm to 1.647 at 425 nm, which is almost parallel to the descending n for the polar tar balls ranging from 1.651 at 365 nm to 1.625 at 425 nm. k is 0.029–0.013 for the nonpolar tar ball over light wavelength of 365–425 nm, while the imaginary part for nonpolar aerosols is 0.007 at 365 nm and zero at wavelength longer than 410 nm, indicating that there is no detectable absorption or k is lower than our detection limit. The overwhelming imaginary part for the nonpolar tar aerosol agrees with many reports that nonpolar or less polar organics have higher absorption compared with the polar BrC in BBOA (Lin et al., 2018; Sengupta et al., 2018). The stronger absorption and relatively higher scattering abilities resulted in a lower SSA compared with the polar tar ball aerosols. The SSA increases towards the visible wavelength from 0.86 at 365 nm to 0.90 at 425 nm for nonpolar tar ball, and the corresponding values are 0.95 to 1.0 for the polar tar balls.
The optical properties of aerosols relate to their chemical composition. Evidently, most of the PAHs identified in the tar ball aerosols with high intensity have strong absorption between 350 and 450 nm (Samburova et al., 2016; Lin et al., 2018), as shown in Fig. S6, which coincide with the range of tar ball absorption measured here, implying that PAHs could be a dominant contributor to the absorption of fresh tar balls. Higher imaginary k can be explained partly by the larger proportion of PAHs as well as more high-molecular-weight organics present in the nonpolar tar ball particles, as conjugated aromatic rings and phenols contribute to the major chromophores in the wood smoke (Laskin et al., 2015; Lin et al., 2017, 2018). High-molecular weight organics may resemble HULIS that can form charge transfer complexes (Phillips and Smith, 2004), and that can absorb light at a longer wavelength range. The result is consistent with the finding that higher molecular weight and aromaticity result in stronger absorption for atmospheric BrC (Dinar et al., 2008). Moreover, the higher NOC content may also contribute to the chromophores in the nonpolar tar aerosols (Lin et al., 2017, 2018).
The average RIs at 375 and 405 nm are 1.671+0.025i and 1.659+0.017i for nonpolar tar ball aerosols. The corresponding RIs are 1.647+0.005i and 1.635+0.04i for the polar tar ball aerosols. The imaginary part k retrieved from the BBCES data, though low, agrees well with k values calculated from UV–visible absorption of the bulk solution. The MAC for the methanol-extracted BrC in tar ball aerosol is shown in Fig. S7. The absorption may be different for complex materials in the particulate and aqueous phases since parameters such as shape factor and mixing state together with artifacts from the optical instruments' detection and data retrieval methods can all affect the final optical results, while solvent-dependent extraction/dissolving efficiency of chromophores or the solvent effect (e.g., pH in water solution) may impact the solution absorption coefficient (Huang et al., 2018; Lin et al., 2017). The light absorption coefficient of particulate BrC has been reported to be 0.7–2.0 times that of bulk BrC extracts by Liu et al. (2013).
Table 2Comparison of tar ball particle optical properties with reference values of BBOA (mean ± standard deviation).
Note: Åabs and Åext were
calculated from tar ball particles with a median diameter of 150 nm in this
study. a Regressed over a wavelength range of 365–400 nm; no
absorption detected over 410 nm using the BBCES system.
b Regressed over a wavelength range of 300–650 nm for bulk fire
plume emissions. c Åabs_UVVIS of methanol
extracts over the whole range from 300/360 to 600 nm. Instrument: IPN
(integrated photoacoustic nephelometer),
CRDS (cavity ring-down
spectrometer), PAS (photoacoustic absorption spectrometer), WELAS (white
light optical particle counter), LWCC (liquid waveguide capillary cell).
The absorption Ångström exponent (Åabs) is often used to describe the wavelength dependence of aerosol light absorption, with a value of nearly 1 for BC particles and values substantially larger than 1 indicating the contribution from BrC (Reid et al., 2005; Chen and Bond, 2010). In this work, Åabs for the nonpolar and polar tar ball particles ranges from 5.9 to 6.8 between 365 and 425 nm, which is consistent with values of 5.7–7.8 calculated from the bulk absorption in solution. The nonpolar tar balls have a lower Åabs. The difference in Åabs reflects the different chemical composition of chromophores in the particles, as inferred also from the AMS data. Bluvshtein et al. (2017) reported relatively low values of Åext (2–3) and Åabs (4–6) over 300–650 nm for ambient fire plume, which are likely affected by BC in the smoke aerosol and are also due to lower wavelength dependence of aerosol absorption and scattering over the longwave visible range. Overall, the broadband optical results for fresh tar ball aerosols are consistent with limited discrete measurements of atmospheric BBOA as summarized in Table 2. It has also been found that the biomass fuel type, combustion conditions, and atmospheric processing eventually affect the optical properties of BBOA. Lack et al. (2012) modeled core-shell absorption for primary organic matter (POM) and BC from biomass burning. They found that the imaginary part of the RI and BrC MAC of POM at 404 nm were 0.007±0.005 and 0.82±0.43 m2 g−1, respectively. Chakrabarty et al. (2010) compared the optical properties of tar balls from smoldering combustion of different biomass. Fuel-dependent imaginary RI for tar ball at 405 nm was 0.008–0.015 and Åabs over 405–532 nm was 4.2–6.4, which is in line with the Åabs value of 6–7 reported for BBOA derived HULIS (Hoffer et al., 2006). Sedlacek et al. (2018) observed a weak absorption for wildfire produced tar balls with RI of 1.56+0.02i. Sumlin et al. (2018) simulated BrC formation under different pyrolysis temperatures. The BrC produced from over 300 ∘C combustion has imaginary part k of 0.05–0.09 and real part n of 1.59–1.68 at 375 nm, and RI at 405 nm is 1.57+0.03i, corresponded Åabs over 375–405 nm is 6.4–7.4.
Optical mixing rules can be used to estimate or explain the refractive indices of internally mixed substances, and three mixing rules are commonly applied in climate models: molar refraction of absorption (Jacobson, 2002), volume-weighted linear average of the refractive indices (d'Almeida et al., 1991), and the Maxwell–Garnett rule (Chýlek et al., 2000). The “linear mixing rule” and molar refraction mixing rules were tested in this work for mixtures of tar ball particles against the retrieved optical data. Relevant data analysis details are provided in the supporting materials (Tables S3–S4, Figs. S8–S12). It was found that both mixing rules can predict the index of refraction for the polar/nonpolar tar balls, and values calculated based on the “linear mixing rule” fit better with the experimental data. As mentioned above, the real fractions for polar and nonpolar BrC contributing to the mass/absorption of BBOA are undefined, some investigations reporting the dynamic polar BrC dominating in mass loading (50 %–85 %) but contributing less to the absorption in BBOA (less than 40 %) (Asa-Awuku et al., 2008; Bluvshtein et al., 2017; Lin et al., 2017, 2018; Rajput et al., 2014; Sengupta et al., 2018). The “linear mixing rule” confirmed in this study should be helpful in the mathematical modeling to assess climatic impacts of biomass burning related BrC aerosol, when their chemical composition is classified.
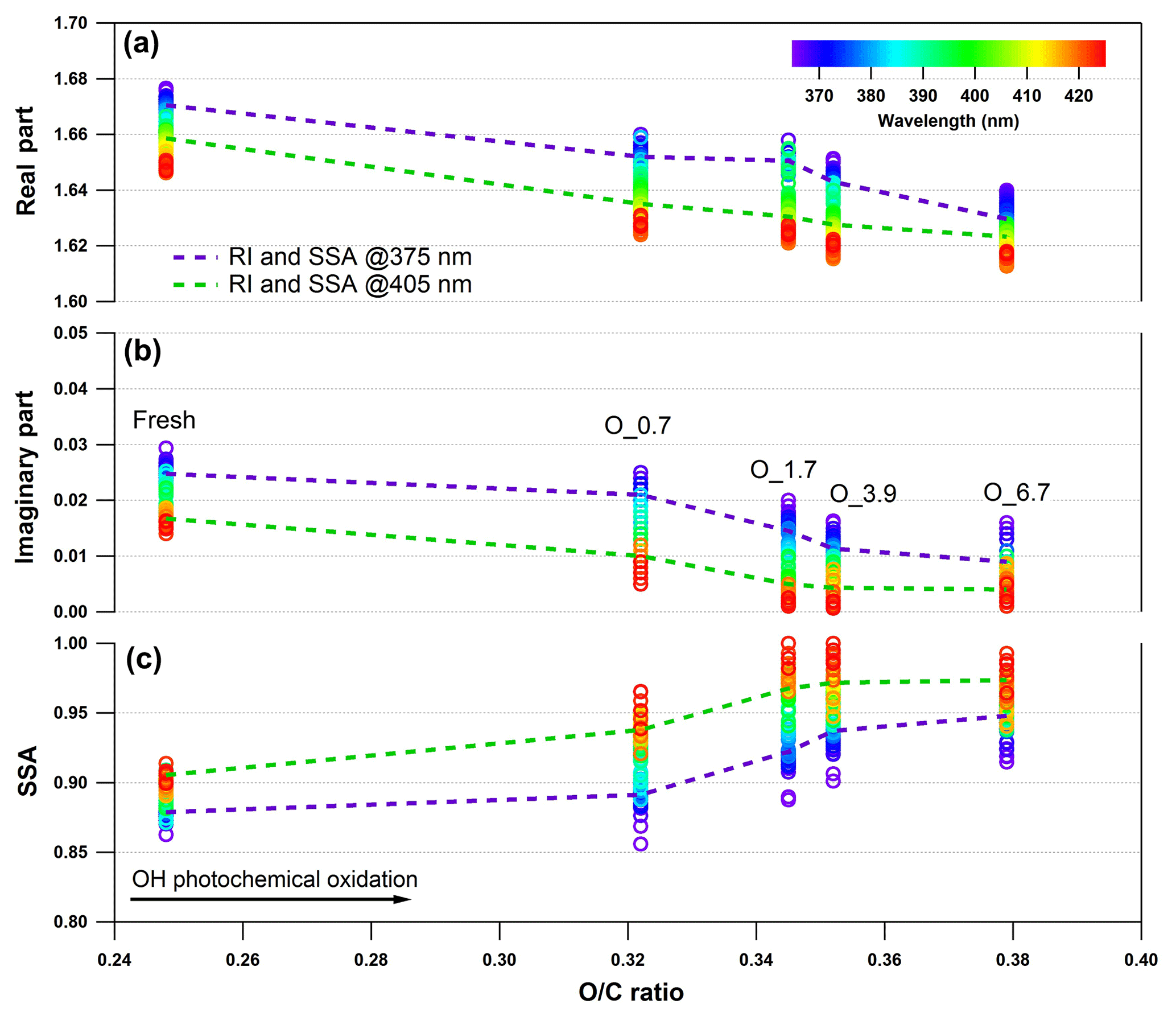
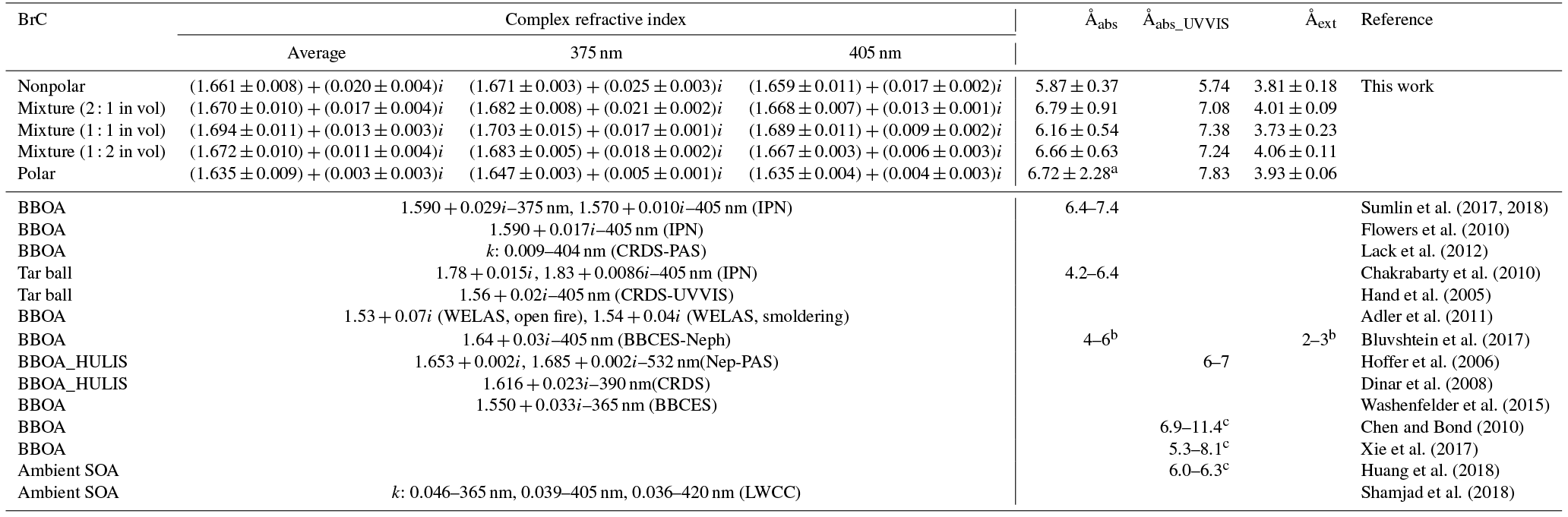
Figure 5Evolution of the retrieved wavelength-dependent complex RI and SSA as a function of O : C ratio for tar ball particles upon OH photochemical oxidation: (a) real part, (b) imaginary part, and (c) SSA calculated for 150 nm particles. The color scale shows the span in the RI for the wavelengths measured from 365 to 425 nm. For clarity, error bars for O : C ratio (±0.01), RI (±0.007 for real part, and ±0.003 for imaginary part on average), and SSA (±0.006) are not shown. Two dashed lines trace the RI and SSA at 375 nm (purple) and 405 nm (green). O_0.7–O_6.7 represent equivalent atmospheric photochemical oxidation for 0.7 and up to 6.7 days.
3.2 Photooxidation of tar ball particles
Aerosols have a wide range of atmospheric lifetimes from hours to days, during which they are involved in various atmospheric processes, resulting in changes in properties (Reid et al., 2005; Rudich et al., 2007; Jimenez et al., 2009). Therefore, we studied the effects of photochemical oxidation of the nonpolar wood-pyrolyzed tar ball aerosols to investigate the physiochemical changes that can occur during tar balls' atmospheric lifetime. Figure 5 presents the evolution of the wavelength-dependent RI and SSA as a function of the aerosols' O : C ratio following NOx-free photochemical aging in the OFR. The oxidation covers 0.7–6.7 EAD. Substantial decrease in the RI and an increase in the SSA are correlated with an increase in the O : C ratio; these specific parameters are summarized in Table S5. Light scattering as well as the absorption by the tar ball aerosol decrease with increasing OH oxidation. The tar aerosols lose their scattering and absorption significantly up to 3.9 EAD aging. The average RI decreased from initial 1.661+0.020i to 1.632+0.007i, and the corresponding average SSA increased from 0.89±0.01 to 0.96±0.02. Then, the RI by tar balls persisted with enhanced oxidation, so that the MAC values remained stable after 3.9 days' oxidation (Fig. S13), suggesting that all the photochemical–labile chromophores were largely eliminated, while the remaining fraction still presented some light absorption. Forrister et al. (2015) also observed a stable fraction of biomass burning BrC that had persistent absorption even after a long photochemical evolution time in the ambient environment. They suggested that the remaining persistent fraction determines the background BrC levels. In our study, the O : C ratio for tar ball aerosols increased continuously with photochemical oxidation, implying production of oxygenated constituents (carboxylic, carbonyl compounds, etc.) and the interaction between these increasingly oxidized species coupled with the relatively stable intrinsic chromophoric structures (e.g., fused aromatic rings in Fig. 3) in some supermolecular structure that may explain the persistent absorption for aged tar ball aerosols (Dewar and Lepley, 1961; Desyaterik et al., 2013; Samburova et al., 2016). In addition, a balance between photobleaching of intrinsic chromophores and photochemical formation of BrC via gas-particle transfer, as well as dynamic gas-particle partitioning of chromophores and products of their photo-degradation, should also be considered in the overall absorption behavior for BBOA during photochemical processes.
The observed photooxidation bleaching is consistent with previous studies on atmospheric processes of BrC. Sumlin et al. (2017) conducted multiple-day photochemical oxidation on primary biomass burning BrC aerosols and observed that BrC loses its light absorption and scattering in the near-UV wavelengths by aging. Their derived RI at 375 nm decreased from 1.59+0.03i for fresh emission to 1.50+0.02i after 4.5 EAD oxidation with a corresponding O : C ratio increase from 0.34 to 0.40. Decrease in the overall BBOA absorption and scattering was also detected in situ following a 1-day evolution by Adler et al. (2011). They monitored an average RI of 1.53+0.07i and 1.54+0.04i for aerosols dominated by open fire and smoldering emissions, respectively, while the RI decreased to 1.49+0.02i of the aged aerosols during the following day. Zhong and Jang (2014) reported that light absorption of wood smoke BrC was modified by the photochemical process, owing to the production of BrC from SOA formation and loss of BrC from photochemical bleaching of the chromophores. The total MAC for the BrC eventually decreased by 19 %–68 % within 1 day of aging. They proposed that bleaching occurred by excitation of electrons through the absorption of sunlight via (UV and near UV illumination) or (visible wavelengths irradiation) transitions. Then, the excited electrons disrupted the conjugated structure of chromophores, leading to the fading of wood smoke color.
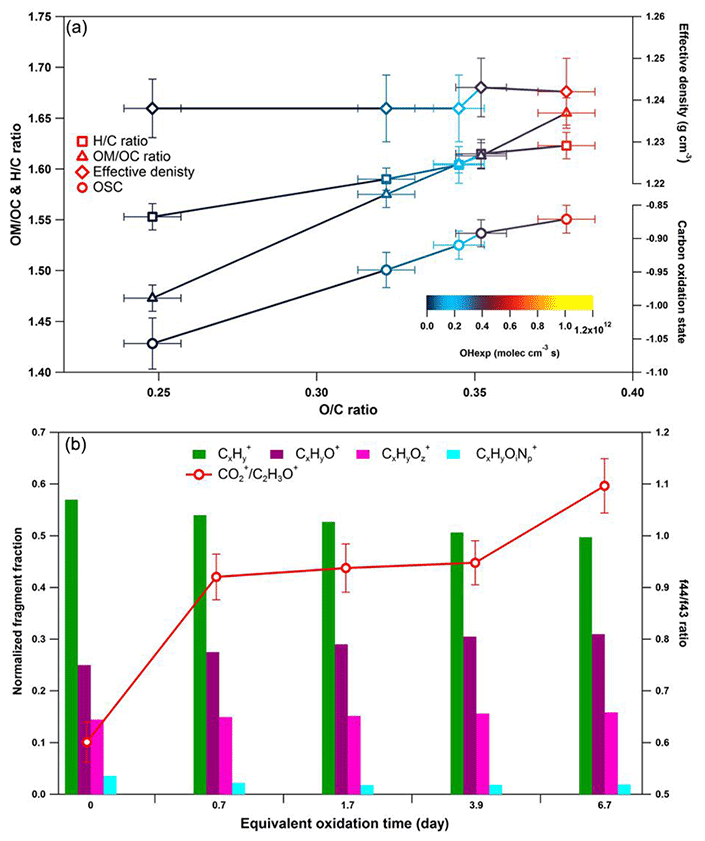
Figure 6Dynamic changes for the chemical characteristics of tar ball particle under NOx-free OH photochemical oxidation: (a) OM ∕ OC, H : C ratio, particle density, and average carbon oxidation state () changes as a function of O : C ratio; (b) mass spectra evolution with oxidation times in term of Cx, CxHyO+, CxHy, and CxHyOi fragment groups.
When tar ball aerosols were illuminated merely by 254 nm UV light at a residence time of 144 s, photolysis occurred and weakly diminished their light absorption, in line with the extent of photon flux exposure. UV irradiation similar to the O_3.9 condition slightly decreased the average RI to 1.649+0.018i, indicating that photolysis played a minor role in tar ball aerosol bleaching and contributed less than 15 % of imaginary k changes in the NOx-free photochemical aging process. Even at full power of UV lamps in the OFR, the average RI decreased by 0.012 and 0.005i for maximum photolyzed tar balls (Tables S6–S7, Figs. S14–S17). As we also did not observe detectable optical changes in blank tests upon exposure to O3 in the dark (Figs. S18–S19), the bleaching of the tar balls in the OFR is mainly attributed to OH-initiated chromophore decomposition via heterogeneous reactions, rather than to O3 oxidation or photolysis.
These results indicate a fundamental relationship between photochemical processes and impairment of light absorbing and scattering abilities in tar ball aerosols. The optical behaviors of tar balls are a consequence of their chemical composition changes, which are very sensitive to photochemical processes, and one daytime atmospheric aging even resulted in significant oxidation and bleaching of tar balls. In Fig. 6a, the H : C, OM ∕ OC, , and particle effective density versus O : C ratios are shown. Figure 6b presents the contributions of Cx, CxHyO+, CxHy, and CxHyOi groups to the tar balls' composition under a range of OH exposure conditions. Mass spectra features and densities of the tar ball aerosols under various oxidation processes are summarized in Table S8. Increasing the OH exposure leads to continuous increase in O : C and H : C ratios, leading to higher for the tar ball aerosols. This result is consistent with Sumlin et al. (2017), who reported that the O : C and H : C for BBOA increased by ∼0.08 and ∼0.03 after 4.5 EAD photochemical oxidation, respectively. In this work, the measured O : C ratio increased from 0.25 to 0.38 after maximum aging, while the H : C ratio increased by 0.07 from an initial value of 1.55.
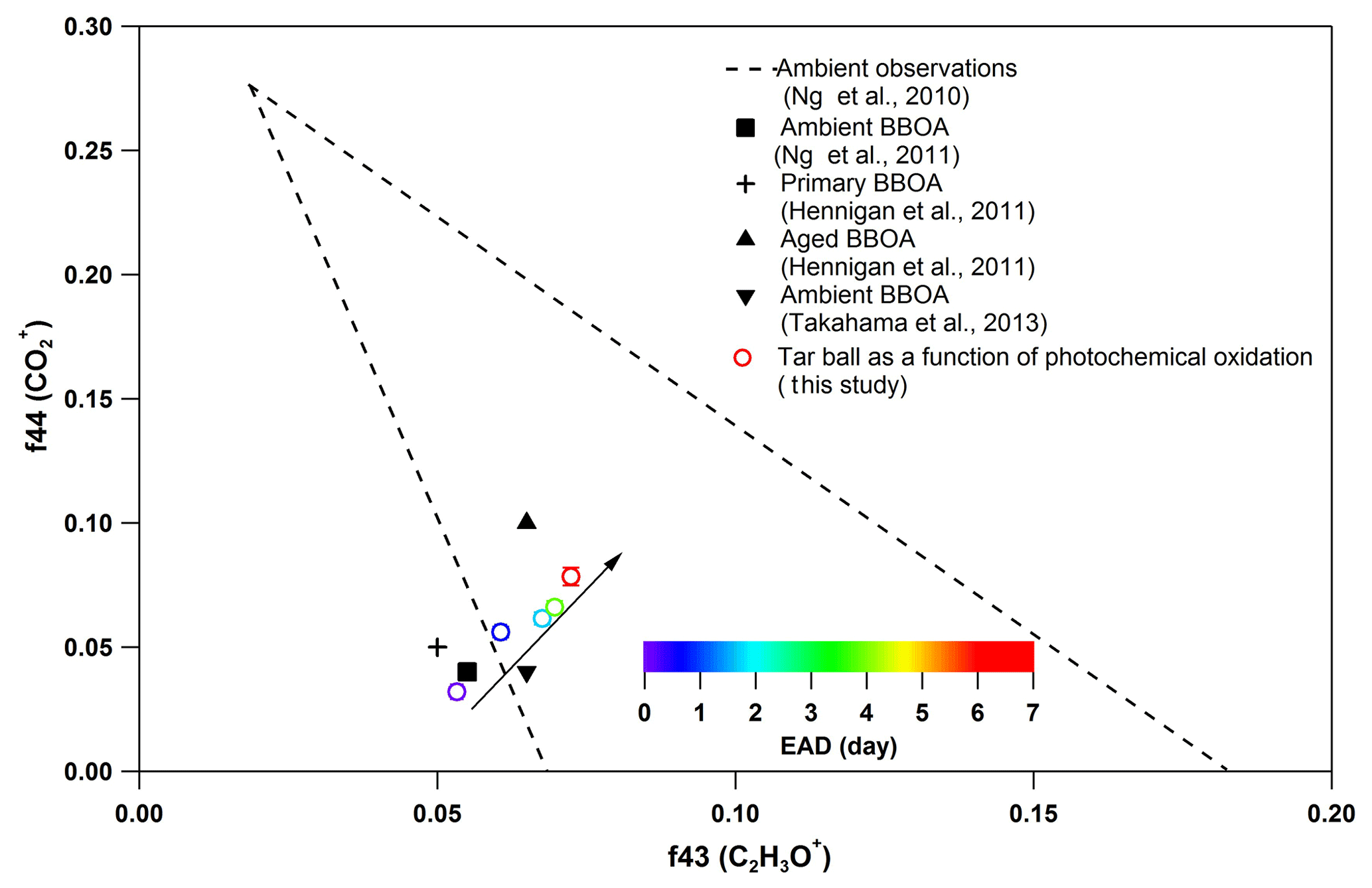
Figure 7Comparison of f44 and f43 values from ambient data sets (Ng et al., 2010) and values from ambient biomass burning organic aerosol.
Other previous studies also depicted a dynamic change in elemental ratios for SOA upon aging (Aiken et al., 2008). The H : C ratio may either increase or decrease, depending on the precursor type and oxidation conditions. Overall, O : C and H : C ratio changes relate to specific chemical processes or/and to gas-particle mass transfer during aging of aerosols (Heald et al., 2010; Kim et al., 2014). The tar ball aerosols consist of mostly reduced species ( < 0), which can be oxidized primarily via oxidative formation of polar functional groups to the carbon skeletons. In OH-initiated oxidation, functionalization includes OH ∕ OOH function group addition and COOH : carbonyl group formation that increase the net oxygen content in SOA (Kroll et al., 2011). Hydration or polar functional group addition to unsaturated C–C bonds may also increase the H : C ratio. Moreover, fragmentation or evaporation also mediates the O : C and H : C ratios of SOA in further aging (Zhang and Seinfeld, 2013; Kim et al., 2014). We attribute the increase in the H : C ratio to such oxidation mechanisms that involve bulk species in the particles. As shown in Table S8, decreased monotonically with aging. After 6.7 EAD photooxidation, contributed only 21 % of the total organic signals. The decrease in indicates that fragmentation reactions are involved in the photochemical evolution, and decomposition of high-molecular weight compounds, thereby, reduced the size of the conjugated molecular system. The persistent high value of after 6.7 EAD photooxidation implies that some high-molecular weight compounds remained in the tar ball aerosols and continue to contribute to light absorption either as individual chromophores or as charge transfer complexes. From Fig. 6b, Cx fragments deplete with OH exposure, while CxHyO+ and CxHy fragments increase, implying the formation of oxygenated moieties in the tar ball aerosols. In addition, a decrease in the CxHyOi fraction was measured from initial 3.6 % to 1.9 % after the maximum oxidation. Ng et al. (2010) suggested using f44() vs. f43(C2H3O+) triangle space as an indication of OA sources and for estimation of their degree of oxidation and volatility. The C2H3O+ (less oxidized fragments) is an indicative fragment from aldehydes or ketones. A high f44∕f43 ratio indicates low volatility and a high oxidation level of SOA. Moreover, high f44∕f43 and O : C ratios are associated with increased hygroscopicity and possible CCN activity of OA (Hennigan et al., 2011; Lambe et al., 2011). The f44 vs. f43 in this study varied with photochemical aging and fell within the expected range for ambient OOA, as shown in Fig. 7. Increase in the f44∕f43 ratio with OH oxidation in Fig. 6b depicted the increase in carboxylic and/or peroxide compounds compared to carbonyl species in the tar balls, which is consistent with the atmospheric evolution of ambient biomass burning plumes (Hennigan et al., 2011; Canonaco et al., 2015).
To infer the possible chemical processes, detailed mass spectra were compared between fresh and 6.7 EAD photochemical oxidized tar balls (Fig. S20). We found that decrease in alkyl/alkenyl chains (e.g., , ), and aromatic ring structure fragments (e.g., , C6) contributed the prominent changes in the Cx group, and a relatively higher increment relative to C2H3O+ explained the increase in the f44∕f43 ratio. The decrease in the abundance of C2H4 (m∕z 60) and C3H5 (m∕z 73) is consistent with recent studies that levoglucosan or similar species can decay in the atmosphere due to photochemical oxidation (Hennigan et al., 2010). The pronounced decrease in intensity at m∕z 137 (C8H9 and C7H5) suggests that the methoxy-phenol components were dissipated substantially in the aged tar balls.
In summary, photochemical oxidation by OH radicals destructed the aromatic rings and methoxy phenolic structures, which are expected to be the primary chromophores in the tar balls. The NOC content and high-molecular weight species were also depleted via OH oxidation. These chemical changes upon OH oxidation may explain the observed diminishing in light scattering and absorption upon photochemical aging.
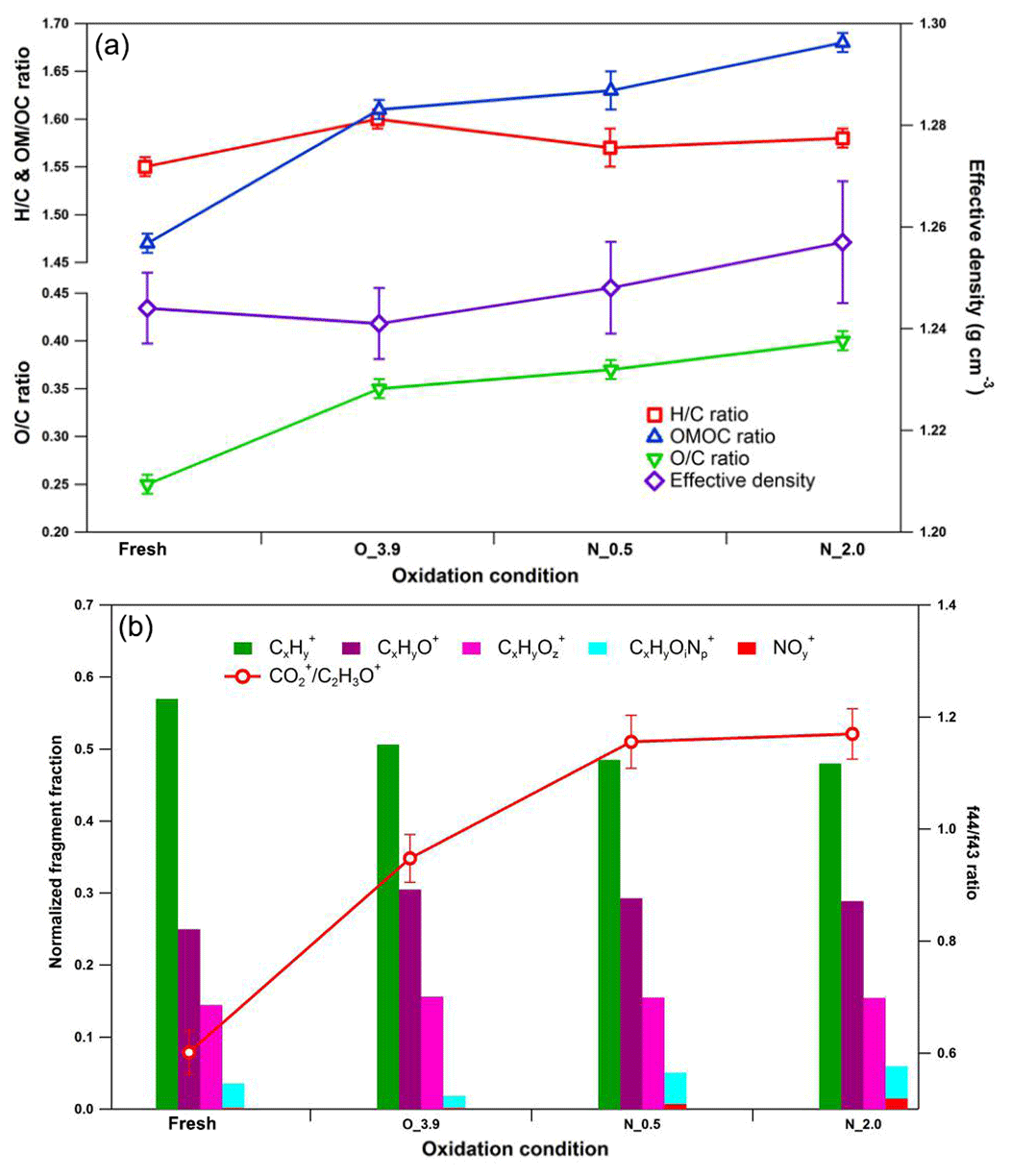
Figure 8Dynamic changes for chemical characteristics of tar ball aerosols under NOx-dependent OH photochemical oxidation: (a) OM ∕ OC, O : C, H : C, and particle density changes; (b) mass spectra changes with different oxidation conditions in terms of Cx, CxHyO+, CxHy, and CxHyOz fragment groups. CxHyOz include all nitrogen-containing fragments (e.g., CxHyON+, CxHyOz, and CxHyN+); include NO+ and . O_3.9 represents 3.9 days' equivalent atmospheric photochemical aging in the absence of NOx; N_0.5 and N_2.0 indicate photochemical oxidation with 0.5 % vol and 2.0 % vol N2O addition at ∼4.0 days' atmospheric oxidation.
3.3 NOx-dependent tar ball particles oxidation
N2O was recently introduced for simulating NOx-dependent SOA formation pathways in OFR under high-O3 concentration, as O(1D)+N2O reactions can be applied to systematically vary the branching ratio of the RO2+NO reactions relative to the RO2+HO2 and/or RO2+RO2 reactions over a range of conditions relevant to atmospheric SOA formation (Lambe et al., 2017). Here we introduced 0.5 % vol and 2.0 % vol N2O to investigate NOx-involved daytime aging of tar ball aerosols in the OFR. The OH exposures were maintained for all these tests at about 4 EAD. The corresponding NO2 concentrations downstream of the OFR were measured to be 96.1±1.3 and 528.3±6.2 ppbv. The concentration of static NO can be neglected under these severe oxidation conditions. Figure 8 shows the mass spectrum features for fresh and aged tar balls reacted in the absence/presence of NOx. Parameters including organic elemental ratios and densities are summarized in Table S8. In general, tar balls oxidized under N2O addition exhibit higher O : C and relatively lower H : C ratios. (NO+ and ) signals appear in the mass spectra and the intensities of display a positive trend with N2O concentration, together with an increase in oxygenated fragments (CxHyO+ and CxHy) and a decrease in hydrocarbon fragments (Cx). The signal ratio of NO+ (m∕z 30) to (m∕z 46) is used to distinguish organic nitrate from inorganic nitrate. The signal from standard inorganic nitrate (e.g., NH4NO3) has a typical ∕ NO+ ratio of 0.485 obtained from our AMS data (detailed mass spectra are shown in Fig. S21). The ratio and standard mass spectra are similar to previous studies (Zhou et al., 2017). The fraction of (NO+ and ) signals in the aged tar balls increased from 0 % to 0.7 % and 1.5 % at 0.5 % vol and 2.0 % vol N2O additions, respectively. The corresponding values of the ∕ NO+ ratio are 0.162 and 0.174, which are much lower than that for inorganic nitrates. Furthermore, the contribution of CxHyOi fragments increased from 1.9 % to 4.4 % and 4.5 % over the course of aging. Therefore, we can conclude that NOC rather than inorganic nitrate formed in the NOx-dependent photooxidation process, resulting in an overall increase in the N : C ratio from 0.010 to 0.012 and 0.015. Additionally, the density of tar balls slightly increased from 1.24 for the fresh tar balls to 1.26 for the one aged in the presence of 2 % vol N2O.
Detailed changes in the mass spectra over the course of the experiment are shown in Fig. S22. Indicative ions of cyclolakyl fragments (e.g., C2, C3, C4) decreased, while NOx addition increased the CO+ and intensities relative to C2H3O+, leading to a higher f44∕f43 ratio. Ng et al. (2007) observed a similar change for photooxidation of terpenes in the presence of NOx. Changes in AMS spectra with NOx addition may mark differences between the dominating reaction pathways in tar ball photooxidation as RO2+NO versus RO2+HO2 and/or RO2+RO2 reactions.
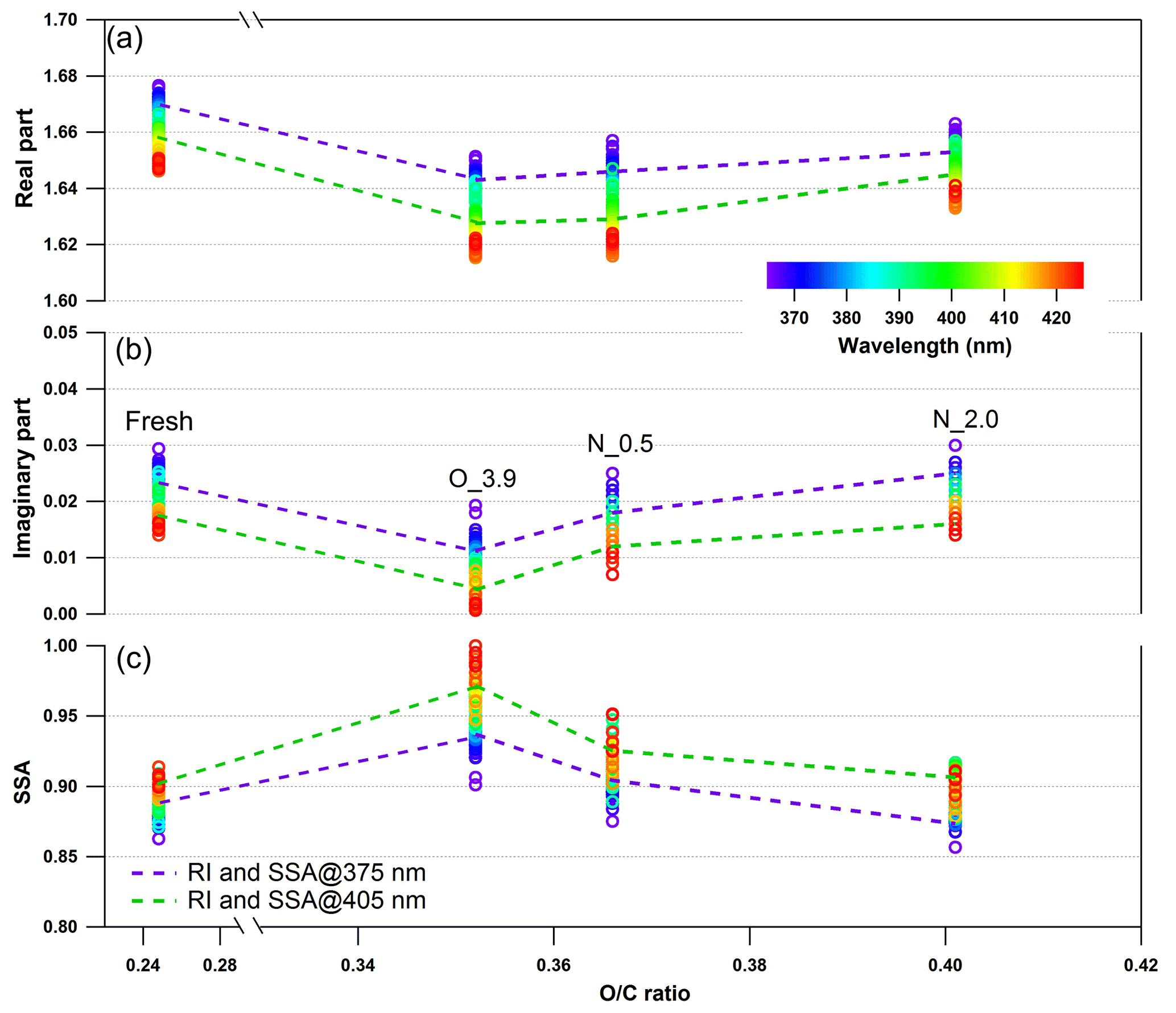
Figure 9Changes in retrieved spectrum-dependent RI as a function of O : C ratio for tar ball particles upon NOx-dependent photochemical oxidation: (a) real part, (b) imaginary part, and (c) SSA calculated from 150 nm particles. For clarity, error bars for O : C ratio (±0.01), RI (±0.006 for real part, and ±0.003 for imaginary part on average), and SSA (±0.007) are not shown. O_3.9 represents 3.9-day equivalent atmospheric photochemical aging in the absence of NOx, N_0.5 and N_2.0 indicate photochemical oxidation with 0.5 and 2.0 % vol N2O addition at ∼4.0 days' atmospheric oxidation.
Photochemical oxidation with NOx addition enhances the oxidation level and increases both the absorption and scattering of tar ball aerosols. Dynamic changes in the complex RI are shown in Fig. 9 and summarized in Table S5. The RI of tar ball aerosols increased from an average of 1.632+0.007i for pure OH-initiated photooxidation to 1.635+0.015i with the addition of 0.5 % vol N2O, and a greater increase up to 1.648+0.019i with 2.0 % vol N2O. The increase in RI is therefore primarily attributed to NOC formation. Zhong and Jang (2014) observed that a higher NOx level slowed photo-bleaching of wood smoke BrC, and they suggested that NOx-modified reaction pathways produce secondary NOC chromophores (i.e., nitro-phenols). Liu et al. (2016) simulated daytime chemistry of various VOCs in the presence of NOx and found that light absorption of produced SOA, especially aromatic ones, increased with NOx concentration. These findings were also corroborated by the experimental study of Lin et al. (2015), where the chemical composition and the light absorption properties of laboratory-generated toluene SOA were reported to have a strong positive dependence on the presence of nitro-phenols formed at high-NOx oxidation conditions. The color of the BrC diminished with photolysis, correlated with a decline of the NOC fraction. Nitration of aromatic species via NOx ∕ N2O5 ∕ NO3 has been proposed as one of the main mechanisms to produce secondary BrC in the atmosphere (Lu et al., 2011; Lin et al., 2015, 2017; Bluvshtein et al., 2017).
The imaginary part at 2.0 % vol N2O addition was almost comparable with that of the fresh tar ball aerosols (average value: RI ), although the real part was lower, suggesting that photooxidation in the presence of NOx promotes the formation of N-containing chromophores via secondary processes. In our experiments, formation of the N-containing chromophores overweighed the bleaching from OH photooxidation to eventually regain the absorption of the aged tar balls. The average SSA calculated for 150 nm particles decreased from 0.96 to 0.91 and 0.89 with N2O addition. Absorption enhancement with N2O addition for tar balls upon photooxidation can also be seen in the MAC changes shown in Fig. S23, where MAC at 375 nm for fresh tar balls was 0.854 m2 g−1; it decreased to 0.416 m2 g−1 via OH photo-bleaching, and then MAC increased to 0.459 m2 g−1 at 0.5 % vol N2O addition, and up to 0.598 m2 g−1 at 2.0 % vol N2O addition due to chromophore formation.
3.4 Atmospheric and climate implication
Atmospheric aging alters the RI of SOA, and the dynamic changes in RI depend on complicated reaction pathways (Liu et al., 2016). OH-initiated photochemical oxidation and photolysis decrease the RI of laboratory proxies of tar balls under NOx-free conditions, while photooxidation under high NOx has an opposite effect on the RI of tar balls. We investigated the relationship between the dynamic RI values of tar ball particles and their possible climatic implications, including the change in light extinction/absorption efficiency and the clear-sky direct radiative forcing. For clarity, light extinction/absorption efficiencies were calculated and compared at wavelengths of 375 and 405 nm, while radiative forcing was estimated over all the measured wavelengths from 365 to 425 nm. Atmospheric and climatic implications were assessed for fresh and oxidized tar balls upon NOx-dependent ∼3.9 EAD photooxidation (O_3.9, N_0.5, and N_2.0), in which fresh tar balls were taken as a reference.
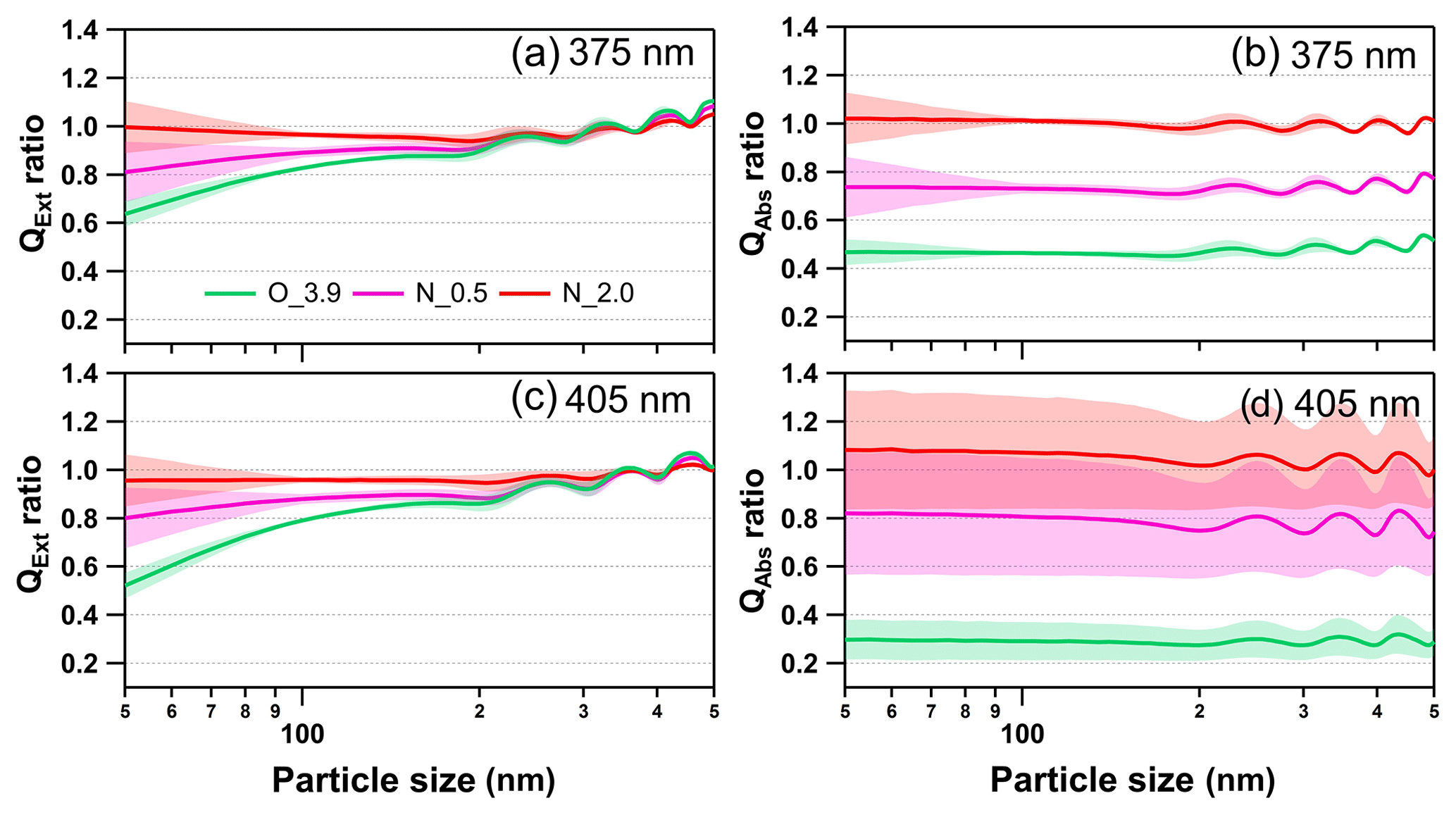
Figure 10Size-resolved light extinction and absorption efficiency ratio of NOx-dependent photooxidized tar balls compared to the fresh tar ball particles: (a) and (c) extinction ratios at 375 and 405 nm; (b) and (d) absorption ratios at 375 and 405 nm. O_3.9 represents 3.9 days' equivalent atmospheric photochemical aging in the absence of NOx; N_0.5 and N_2.0 indicate photochemical oxidation with 0.5 % vol and 2.0 % vol N2O addition at ∼4.0 days' atmospheric oxidation.
As shown in Fig. 10, photochemical oxidation under NOx-free condition (O_3.9) diminished light extinction and absorption efficiency of tar ball aerosols in the atmospheric relevant size of 50–300 nm, causing about 5 %–40 % decrease in extinction at 375 and 405 nm wavelength. For aerosols larger than 400 nm, the extinction efficiency of tar ball aerosols increased instead after photochemical aging. The light extinction efficiency presented higher size-dependence than absorption, and extinction changes were more sensitive to particle size, especially in the smaller sizes. The decreased absorption was more pronounced, with an ∼60 % decrease at 375 nm and over 75 % at 405 nm. Previous studies have confirmed the relationship between biomass burning emissions and acute regional visibility deterioration (Huang et al., 2012; Chen et al., 2017). Our results demonstrate that OH radical initiated daytime aging may play an important role in improving visibility degradation caused by primary biomass BrC. However, photochemical evolution under high-NOx conditions may compensate effects of the photooxidation bleaching of tar ball aerosols via the formation of NOC chromophores. At N_0.5 conditions, the light extinction decreased by 4 % to 20 % at 375 nm and 5 % to 24 % at 405 nm, respectively. The corresponding absorption decrease was 20 %–27 % at both wavelengths. With more N2O addition, formation of secondary N-containing chromophores almost completely offsets light extinction/absorption decrease caused by photooxidation. Under the N_2.0 conditions, enhancement of light absorption efficiency for tar ball was about 0 %–9 % at 405 nm in the entire size range of 50–500 nm.
Radiative forcing from aerosols over both ground and snow is vital to climate models (Barnett et al., 2005; Kanakidou et al., 2005). Integrated radiative forcing for tar ball aerosols as a function of particle size under various oxidation conditions is shown in Fig. 11. Size-/wavelength-resolved SRF are also shown in Figs. S24 and S25. Integrated SRF over ground has negative values for tar balls over almost all the atmospheric relevant sizes, indicating a radiative cooling effect by tar ball aerosols except at 195–210 nm, where fresh tar ball particles present warming effect with SRF up to ∼0.48 W g−1. In practical fire emissions, the size of tar balls depends on the burning and environment conditions and biomass fuel types with typical values between tens to hundreds of nanometers (Reid et al., 2005; Pósfai et al., 2004). The complicated size-dependence character of SRF makes it difficult to assess the real climatic effect of tar ball particles without extensive calculations. Figure 11a suggests fresh tar balls have SRF values of −7.46 W g−1 at 150 nm and 0.45 W g−1 at 200 nm, respectively. The SRF decreased for all size ranges due to photochemical oxidation to −7.93 W g−1 at 150 nm and −1.37 W g−1 at 200 nm for tar ball aerosols under O_3.9 condition. At N_0.5 conditions, SRF was −7.37 W g−1 at 150 nm and 0.16 W g−1 at 200 nm, and the corresponding values at N_2.0 conditions increased to −7.20 W g−1 at 150 nm and 0.31 W g−1 at 200 nm.
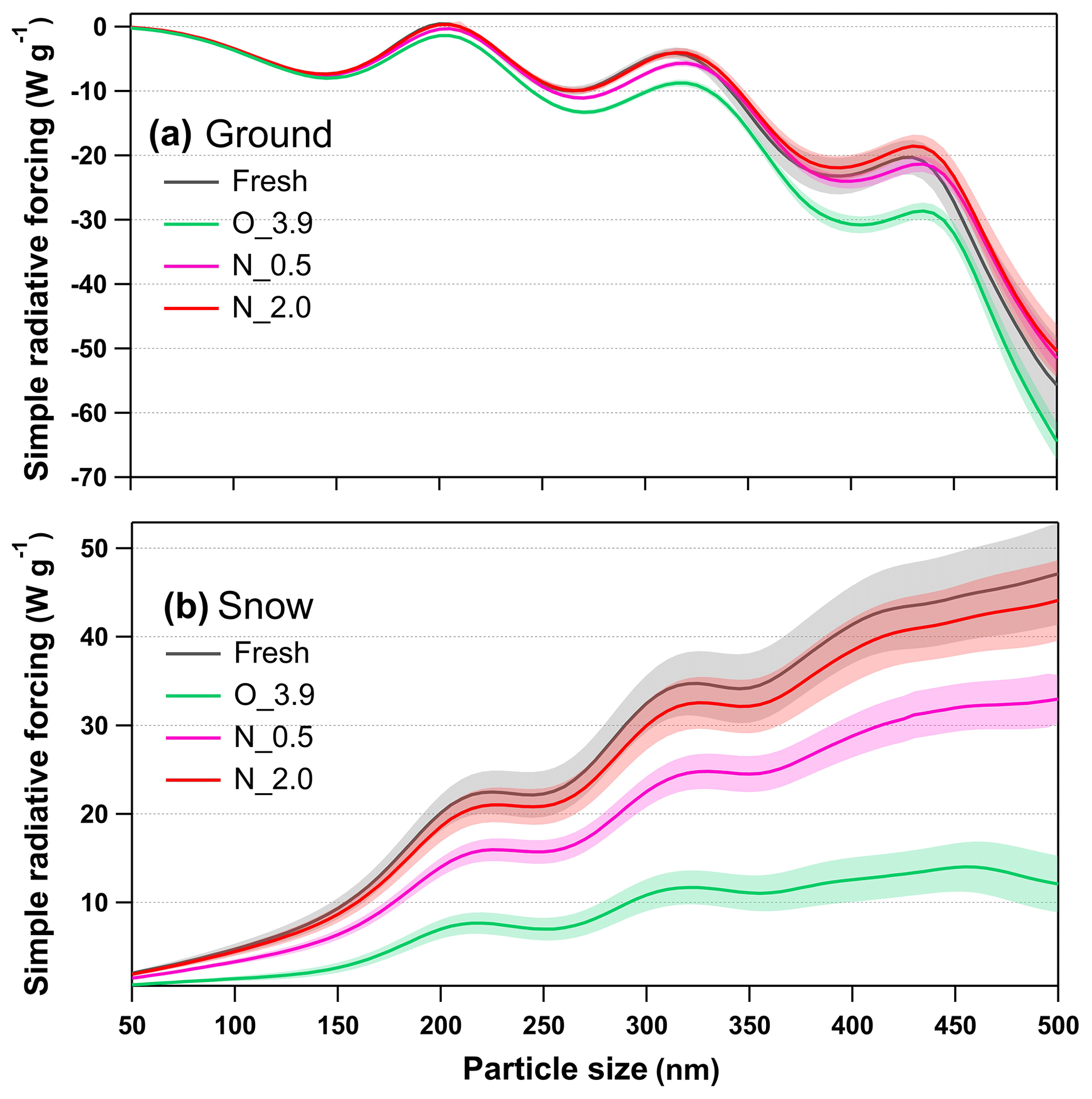
Figure 11Calculated size-resolved simple radiative forcing (SRF, W g−1) by tar ball aerosols, integrated over 365–425 nm incident solar irradiation for fresh and NOx-dependent photooxidized tar balls: (a) ground-based radiative forcing, (b) snow-based radiative forcing.
In contrast, tar ball particles contributed to positive forcing (warming effect) over the bright terrain throughout the atmospheric aging, as shown in Fig. 11b. Radiative forcing over the snow showed a simple increasing trend with particle size, indicating that larger BrC aerosol with identical mass loading in the air have a higher warming effect. The changes in snow-based radiative forcing upon photochemical aging followed the same trends as in the ground-based cases. Fresh tar ball at size of 200 nm has SRF of 20.12 W g−1 over the incident solar wavelength of 365–425 nm on the snow terrain. With photochemical oxidation under NOx-free condition, radiative forcing decreased significantly. After 3.9 EAD atmospheric aging, snow-based radiative forcing for tar ball decreased by 65 %–73 % over the size range of 50–500 nm, the value of 200 nm tar ball became 6.99 W g−1. When NOx was involved in the photochemical oxidation of tar balls, the decrement of radiative forcing was weakened. At N_0.5, SRF for 200 nm tar ball was 14.01 W g−1, while at N_2.0 condition, size-dependent SRF from the aged tar ball was almost comparable with that from fresh tar ball, and SRF for 200 nm tar ball was 18.56 W g−1.
Although less than 10 % of the solar spectrum's energy is distributed between 365 and 425 nm, the radiative forcing over this range represents a significant warming or cooling potential over the arctic terrain. In conclusion, photochemical oxidation under NOx-free conditions can decrease radiative forcing of tar ball aerosols, resulting in enhancement in the cooling effect over ground and decreased in warming effect over the snow. However, NOx involvement in photooxidation inhibits the decrease in radiative forcing of tar ball aerosols. Overall, the complex changes in optical properties of tar balls at long aging times impose great uncertainties in traditional model-based estimation of BBOA. Our study emphasizes the importance of taking this atmospheric process into consideration to refine the understanding of the climatic and atmospheric influences from these aerosols.
In this study, proxies for tar ball aerosols were generated in the laboratory following a flameless wood pyrolysis process. The optical and chemical properties of the generated tar balls were constrained using BBCES and HR-Tof-AMS/SP-LD-REMPI-MS and were shown to have many similarities to ambient biomass burning aerosols. Laboratory-generated fresh tar ball aerosols have light absorption characteristics similar to atmospheric BrC with higher absorption efficiency towards the UV. The average complex refractive indices between 365 and 425 nm are 1.661+0.020i and 1.635+0.003i for nonpolar and polar tar ball aerosols, respectively.
Atmospheric evolution for tar ball aerosols was experimentally simulated using an oxidation flow reactor. The study focused on dynamic changes in the optical and chemical properties due to NOx-dependent photochemical oxidation. Furthermore, the relationship between oxidation level and the resulting RI of the tar ball aerosols was explored. We found a substantial decrease in the scattering and absorption properties of tar balls, with a corresponding increase in SSA with OH oxidation in the absence of NOx. A correlation between the RI decrease and increase in the O : C and H : C ratios was observed. The decrease in light scattering and absorption is attributed to the destruction of aromatic/phenolic/NOC and high-molecular weight species chromophores via OH-initiated photooxidation of tar balls. Over longer aging times, the average RI of the tar ball aerosols decreased from 1.661+0.020i to 1.632+0.007i upon atmospheric equivalent to 3.9 days' aging, and the corresponding O : C and H : C ratio increased from an initial 0.25 and 1.55 to 0.35 and 1.59, respectively.
Our results suggest that OH oxidation rather than photolysis or ozone reactions plays the dominant role that determines the optical and chemical properties in tar ball aging. The observed decrease in absorption results from depletion of chromophores such as aromatic rings, phenolic compounds and high-molecular weight species.
Simulations under high-NOx environments enhanced the aerosol oxidation state and increased the scattering and absorption of tar ball aerosols relative to OH photooxidation in the absence of NOx. At ∼3.9 EAD, addition of 0.5 % vol and 2.0 % vol N2O increased the organic elemental ratios (O : C, H : C, and N : C ratios) and doubled the organic nitrate fraction in the particles from 1.9 % to ∼4.4 %. The formation of NOC chromophores overweighs the intrinsic depletion of chromophores, leading to a higher RI of 1.635+0.015i and 1.648+0.019i.
The atmospheric and climatic implications from tar ball aerosols under various oxidation conditions were assessed using a simple radiative forcing model in terms of extinction/absorption efficiency changes and ground-/snow-based radiative forcing. These results demonstrate that the optical and chemical properties of tar ball particles are dynamically related to atmospheric aging, and optical changes are governed by both photobleaching and secondary chromophore formation. Therefore, the atmospheric process should be emphasized in model predictions for evaluating biomass burning BrC aerosol radiative forcing as well as climate change.
Data are publicly available from the public website of the Weizmann Institute of Science (https://weizmann.alma.exlibrisgroup.com/view/delivery/972WIS_INST/1245276980003596, Rudich, 2018).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-139-2019-supplement.
YR was the project leader. YR, CL, and QF designed the experiment, and CL and QF conducted the experiment. JS, JP, and RZ contributed the REMPI-MS measurement and revised related paragraphs in the manuscript. DM improved the code in the SRF calculation. CL wrote the manuscript with the contribution and help of LA.
The authors declare that they have no conflict of interest.
This research was partially supported by research grants from US–Israel
Binational Science Foundation (BSF) grant no. 2016093 and the Israel Ministry
of Science, Maimonide program. Chunlin Li acknowledges support from the
Planning & Budgeting Committee, Israel (2018/19). Julian Schade,
Johannes Passig, and Ralf Zimmermann
gratefully acknowledge financial support from the German Research Foundation,
project number ZI 764/6-1, and Photonion GmbH, Schwerin,
Germany.
Edited by: Ryan Sullivan
Reviewed by: two anonymous referees
Abo Riziq, A., Erlick, C., Dinar, E., and Rudich, Y.: Optical properties of absorbing and non-absorbing aerosols retrieved by cavity ring down (CRD) spectroscopy, Atmos. Chem. Phys., 7, 1523–1536, https://doi.org/10.5194/acp-7-1523-2007, 2007.
Adachi, K. and Buseck, P. R.: Atmospheric tar balls from biomass burning in Mexico, J. Geophys. Res.-Atmos., 116, D05204, https://doi.org/10.1029/2010JD015102, 2011.
Adler, G., Riziq, A. A., Erlick, C., and Rudich, Y.: Effect of intrinsic organic carbon on the optical properties of fresh diesel soot, Proc. Natl. Acad. Sci. USA, 107, 6699–6704, https://doi.org/10.1073/pnas.0903311106, 2010.
Adler, G., Flores, J. M., Abo Riziq, A., Borrmann, S., and Rudich, Y.: Chemical, physical, and optical evolution of biomass burning aerosols: a case study, Atmos. Chem. Phys., 11, 1491–1503, https://doi.org/10.5194/acp-11-1491-2011, 2011.
Aiken, A. C., Decarlo, P. F., Kroll, J. H., Worsnop, D. R., Huffman, J. A., Docherty, K. S., Ulbrich, I. M., Mohr, C., Kimmel, J. R., and Sueper, D.: O ∕ C and OM ∕ OC ratios of primary, secondary, and ambient organic aerosols with high-resolution time-of-flight aerosol mass spectrometry, Environ. Sci. Technol., 42, 4478–4485, https://doi.org/10.1021/es703009q, 2008.
Alexander, D. T., Crozier, P. A., and Anderson, J. R.: Brown carbon spheres in East Asian outflow and their optical properties, Science, 321, 833–836, https://doi.org/10.1126/science.1155296, 2008.
Andreae, M. O. and Gelencsér, A.: Black carbon or brown carbon? The nature of light-absorbing carbonaceous aerosols, Atmos. Chem. Phys., 6, 3131–3148, https://doi.org/10.5194/acp-6-3131-2006, 2006.
Asa-Awuku, A., Sullivan, A. P., Hennigan, C. J., Weber, R. J., and Nenes, A.: Investigation of molar volume and surfactant characteristics of water-soluble organic compounds in biomass burning aerosol, Atmos. Chem. Phys., 8, 799–812, https://doi.org/10.5194/acp-8-799-2008, 2008.
Bahadur, R., Praveen, P. S., Xu, Y., and Ramanathan, V.: Solar absorption by elemental and brown carbon determined from spectral observations, Proc. Natl. Acad. Sci. USA, 109, 17366–17371, https://doi.org/10.1073/pnas.1205910109, 2012.
Barnett, T. P., Adam, J. C., and Lettenmaier, D. P.: Potential impacts of a warming climate on water availability in snow-dominated regions, Nature, 438, 303–309, https://doi.org/10.1038/nature04141, 2005.
Bente, M., Sklorz, M., Streibel, T., and Zimmermann, R.: Online laser desorption-multiphoton postionization mass spectrometry of individual aerosol particles: molecular source indicators for particles emitted from different traffic-related and wood combustion sources, Anal. Chem., 80, 8991–9004, https://doi.org/10.1021/ac801295f, 2008.
Bente, M., Sklorz, M., Streibel, T., and Zimmermann, R.: Thermal desorption-multiphoton ionization time-of-flight mass spectrometry of individual aerosol particles: A simplified approach for online single-particle analysis of Polycyclic Aromatic Hydrocarbons and their derivatives, Anal. Chem., 81, 2525–2536, https://doi.org/10.1021/ac802296f, 2009.
Bluvshtein, N., Lin, P., Flores, J. M., Segev, L., Mazar, Y., Tas, E., Snider, G., Weagle, C., Brown, S. S., and Laskin, A.: Broadband optical properties of biomass-burning aerosol and identification of brown carbon chromophores, J. Geophys. Res.-Atmos. 122, 5441–5456, https://doi.org/10.1002/2016JD026230, 2017.
Boesl, U., Neusser, H., and Schlag, E.: Two-photon ionization of polyatomic molecules in a mass spectrometer, Z. Naturforsch. A, 33, 1546–1548, https://doi.org/10.1515/zna-1978-1218, 1978.
Bond, T. C. and Bergstrom, R. W.: Light absorption by carbonaceous particles: An investigative review, Aerosol Sci. Tech., 40, 27–67, https://doi.org/10.1080/02786820500421521, 2006.
Canonaco, F., Slowik, J. G., Baltensperger, U., and Prévôt, A. S. H.: Seasonal differences in oxygenated organic aerosol composition: implications for emissions sources and factor analysis, Atmos. Chem. Phys., 15, 6993–7002, https://doi.org/10.5194/acp-15-6993-2015, 2015.
Chakrabarty, R. K., Moosmüller, H., Chen, L.-W. A., Lewis, K., Arnott, W. P., Mazzoleni, C., Dubey, M. K., Wold, C. E., Hao, W. M., and Kreidenweis, S. M.: Brown carbon in tar balls from smoldering biomass combustion, Atmos. Chem. Phys., 10, 6363–6370, https://doi.org/10.5194/acp-10-6363-2010, 2010.
Chen, Y. and Bond, T. C.: Light absorption by organic carbon from wood combustion, Atmos. Chem. Phys., 10, 1773–1787, https://doi.org/10.5194/acp-10-1773-2010, 2010.
Chen, J., Li, C., Ristovski, Z., Milic, A., Gu, Y., Islam, M. S., Wang, S., Hao, J., Zhang, H., and He, C.: A review of biomass burning: Emissions and impacts on air quality, health and climate in China, Sci. Total Environ., 579, 1000–1034, https://doi.org/10.1016/j.scitotenv.2016.11.025, 2017.
Chung, C. E., Ramanathan, V., and Decremer, D.: Observationally constrained estimates of carbonaceous aerosol radiative forcing, P. Natl. Acad. Sci. USA,, 109, 11624–11629, https://doi.org/10.1073/pnas.1203707109, 2012.
Chýlek, P., Videen, G., Geldart, D., Dobbie, J. S., and Tso, H.: Effective medium approximations for heterogeneous particles, Light scattering by nonspherical particles: theory, measurements, and applications, 273–308, edited by: Mishchenko, M. I., Hovenier, J. W., and Travis, L. D., Academic Press, San Diego, 2000.
Czech, H., Pieber, S. M., Tiitta, P., Sippula, O., Kortelainen, M., Lamberg, H., Grigonyte, J., Streibel, T., Prévôt, A. S., and Jokiniemi, J.: Time-resolved analysis of primary volatile emissions and secondary aerosol formation potential from a small-scale pellet boiler, Atmos. Environ., 158, 236–245, https://doi.org/10.1016/j.atmosenv.2017.03.040, 2017.
d'Almeida, G. A., Koepke, P., and Shettle, E. P.: Atmospheric aerosols: global climatology and radiative characteristics, A. Deepak Publishing, Hampton, Va, 1991.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., and Docherty, K. S.: Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
Desyaterik, Y., Sun, Y., Shen, X., Lee, T., Wang, X., Wang, T., and Collett, J. L.: Speciation of “brown” carbon in cloud water impacted by agricultural biomass burning in eastern China, J. Geophys. Res.-Atmos., 118, 7389–7399, https://doi.org/10.1002/jgrd.50561, 2013.
Dewar, M. J. and Lepley, A. R.: π-Complexes. I. Charge Transfer Spectra of π-Complexes Formed by Trinitrobenzene with Polycyclic Aromatic Compounds, J. Am. Chem. Soc., 83, 4560–4563, 1961.
Dinar, E., Riziq, A. A., Spindler, C., Erlick, C., Kiss, G., and Rudich, Y.: The complex refractive index of atmospheric and model humic-like substances (HULIS) retrieved by a cavity ring down aerosol spectrometer (CRD-AS), Faraday Discuss., 137, 279–295, https://doi.org/10.1039/B703111D, 2008.
Epstein, S. A., Blair, S. L., and Nizkorodov, S. A.: Direct photolysis of α-pinene ozonolysis secondary organic aerosol: effect on particle mass and peroxide content, Environ. Sci. Technol., 48, 11251–11258, https://doi.org/10.1021/es502350u, 2014.
Feng, Y., Ramanathan, V., and Kotamarthi, V. R.: Brown carbon: a significant atmospheric absorber of solar radiation?, Atmos. Chem. Phys., 13, 8607–8621, https://doi.org/10.5194/acp-13-8607-2013, 2013.
Finewax, Z., de Gouw, J. A., and Ziemann, P. J.: Identification and quantification of 4-Nitrocatechol formed from OH and NO3 radical-initiated reactions of catechol in air in the presence of NOx: Implications for secondary organic aerosol formation from biomass burning, Environ. Sci. Technol., 52, 1981–1989, https://doi.org/10.1021/acs.est.7b05864, 2018.
Flores, J. M., Zhao, D. F., Segev, L., Schlag, P., Kiendler-Scharr, A., Fuchs, H., Watne, Å. K., Bluvshtein, N., Mentel, Th. F., Hallquist, M., and Rudich, Y.: Evolution of the complex refractive index in the UV spectral region in ageing secondary organic aerosol, Atmos. Chem. Phys., 14, 5793–5806, https://doi.org/10.5194/acp-14-5793-2014, 2014a.
Flores, J. M., Washenfelder, R., Adler, G., Lee, H., Segev, L., Laskin, J., Laskin, A., Nizkorodov, S., Brown, S., and Rudich, Y.: Complex refractive indices in the near-ultraviolet spectral region of biogenic secondary organic aerosol aged with ammonia, Phys. Chem. Chem. Phys., 16, 10629–10642, https://doi.org/10.1039/C4CP01009D, 2014b.
Flowers, B. A., Dubey, M. K., Mazzoleni, C., Stone, E. A., Schauer, J. J., Kim, S.-W., and Yoon, S. C.: Optical-chemical-microphysical relationships and closure studies for mixed carbonaceous aerosols observed at Jeju Island; 3-laser photoacoustic spectrometer, particle sizing, and filter analysis, Atmos. Chem. Phys., 10, 10387–10398, https://doi.org/10.5194/acp-10-10387-2010, 2010.
Forrister, H., Liu, J., Scheuer, E., Dibb, J., Ziemba, L., Thornhill, K. L., Anderson, B., Diskin, G., Perring, A. E., and Schwarz, J. P.: Evolution of brown carbon in wildfire plumes, Geophys. Res. Lett., 42, 4623–4630, https://doi.org/10.1002/2015GL063897, 2015.
Forster, P. M. F. and Taylor, K. E.: Climate forcings and climate sensitivities diagnosed from coupled climate model integrations, J. Climate, 19, 6181–6194, https://doi.org/10.1175/JCLI3974.1, 2006.
Fu, H., Zhang, M., Li, W., Chen, J., Wang, L., Quan, X., and Wang, W.: Morphology, composition and mixing state of individual carbonaceous aerosol in urban Shanghai, Atmos. Chem. Phys., 12, 693–707, https://doi.org/10.5194/acp-12-693-2012, 2012.
Ge, X., Setyan, A., Sun, Y., and Zhang, Q.: Primary and secondary organic aerosols in Fresno, California during wintertime: Results from high resolution aerosol mass spectrometry, J. Geophys. Res.-Atmos., 117, D19301, https://doi.org/10.1029/2012JD018026, 2012.
Grotemeyer, J., Boesl, U., Walter, K., and Schlag, E. W.: A general soft ionization method for mass spectrometry: Resonance-enhanced multi-photon ionization of biomolecules, Org. Mass. Spectrom., 21, 645–653, https://doi.org/10.1002/oms.1210211008, 1986.
Hand, J. L., Malm, W., Laskin, A., Day, D., Lee, T. B., Wang, C., Carrico, C., Carrillo, J., Cowin, J. P., and Collett, J.: Optical, physical, and chemical properties of tar balls observed during the Yosemite Aerosol Characterization Study, J. Geophys. Res.-Atmos., 110, D21210, https://doi.org/10.1029/2004JD005728, 2005.
He, L.-Y., Lin, Y., Huang, X.-F., Guo, S., Xue, L., Su, Q., Hu, M., Luan, S.-J., and Zhang, Y.-H.: Characterization of high-resolution aerosol mass spectra of primary organic aerosol emissions from Chinese cooking and biomass burning, Atmos. Chem. Phys., 10, 11535–11543, https://doi.org/10.5194/acp-10-11535-2010, 2010.
He, Q., Bluvshtein, N., Segev, L., Meidan, D., Flores, J. M., Brown, S. S., Brune, W., and Rudich, Y.: Evolution of the complex refractive index of secondary organic aerosols during atmospheric aging, Environ. Sci. Technol., 52, 3456–3465, https://doi.org/10.1021/acs.est.7b05742, 2018.
Heald, C., Kroll, J., Jimenez, J., Docherty, K., DeCarlo, P., Aiken, A., Chen, Q., Martin, S., Farmer, D., and Artaxo, P.: A simplified description of the evolution of organic aerosol composition in the atmosphere, Geophys. Res. Lett., 37, L08803, https://doi.org/10.1029/2010GL042737, 2010.
Hennigan, C. J., Sullivan, A. P., Collett Jr., J. L., and Robinson, A. L.: Levoglucosan stability in biomass burning particles exposed to hydroxyl radicals, Geophys. Res. Lett., 37, L09806, https://doi.org/10.1029/2010GL043088, 2010.
Hennigan, C. J., Miracolo, M. A., Engelhart, G. J., May, A. A., Presto, A. A., Lee, T., Sullivan, A. P., McMeeking, G. R., Coe, H., Wold, C. E., Hao, W.-M., Gilman, J. B., Kuster, W. C., de Gouw, J., Schichtel, B. A., Collett Jr., J. L., Kreidenweis, S. M., and Robinson, A. L.: Chemical and physical transformations of organic aerosol from the photo-oxidation of open biomass burning emissions in an environmental chamber, Atmos. Chem. Phys., 11, 7669–7686, https://doi.org/10.5194/acp-11-7669-2011, 2011.
Heger, H. J., Zimmermann, R., Dorfner, R., Beckmann, M., Griebel, H., Kettrup, A., and Boesl, U.: On-line emission analysis of polycyclic aromatic hydrocarbons down to pptv concentration levels in the flue gas of an incineration pilot plant with a mobile resonance-enhanced multiphoton ionization time-of-flight mass spectrometer, Anal. Chem., 71, 46–57, https://doi.org/10.1021/ac980611y, 1999.
Herring, C. L., Faiola, C. L., Massoli, P., Sueper, D., Erickson, M. H., McDonald, J. D., Simpson, C. D., Yost, M. G., Jobson, B. T., and VanReken, T. M.: New methodology for quantifying polycyclic aromatic hydrocarbons (PAHs) using high-resolution aerosol mass spectrometry, Aerosol Sci. Tech., 49, 1131–1148, https://doi.org/10.1080/02786826.2015.1101050, 2015.
Hoffer, A., Gelencsér, A., Guyon, P., Kiss, G., Schmid, O., Frank, G. P., Artaxo, P., and Andreae, M. O.: Optical properties of humic-like substances (HULIS) in biomass-burning aerosols, Atmos. Chem. Phys., 6, 3563–3570, https://doi.org/10.5194/acp-6-3563-2006, 2006.
Hoffer, A., Tóth, A., Nyiro-Kósa, I., Pósfai, M., and Gelencsér, A.: Light absorption properties of laboratory-generated tar ball particles, Atmos. Chem. Phys., 16, 239–246, https://doi.org/10.5194/acp-16-239-2016, 2016.
Huang, K., Zhuang, G., Lin, Y., Fu, J. S., Wang, Q., Liu, T., Zhang, R., Jiang, Y., Deng, C., Fu, Q., Hsu, N. C., and Cao, B.: Typical types and formation mechanisms of haze in an Eastern Asia megacity, Shanghai, Atmos. Chem. Phys., 12, 105–124, https://doi.org/10.5194/acp-12-105-2012, 2012.
Huang, R. J., Yang, L., Cao, J. J., Chen, Y., Chen, Q., Li, Y., Duan, J., Zhu, C., Dai, W., and Wang, K.: Brown carbon aerosol in Urban Xi'an, Northwest China: the composition and light absorption properties, Environ. Sci. Technol., 52, 6825–6833, https://doi.org/10.1021/acs.est.8b02386, 2018.
IPCC: Climate Change 2013: the physical science basis. Contribution of working group I to the fifth assessment report of the intergovernmental panel on climate change, Cambridge, and New York, NY, Cambridge University Press, 2013.
Jacobson, M. Z.: Analysis of aerosol interactions with numerical techniques for solving coagulation, nucleation, condensation, dissolution, and reversible chemistry among multiple size distributions, J. Geophys. Res.-Atmos., 107, 4366, https://doi.org/10.1029/2001JD002044, 2002.
Jacobson, M. Z.: Investigating cloud absorption effects: Global absorption properties of black carbon, tar balls, and soil dust in clouds and aerosols, J. Geophys. Res.-Atmos., 117, https://doi.org/10.1029/2011JD017218, 2012.
Jacobson, M. Z.: Effects of biomass burning on climate, accounting for heat and moisture fluxes, black and brown carbon, and cloud absorption effects, J. Geophys. Res.-Atmos., 119, 8980–9002, doi.org/10.1002/2014JD021861, 2014.
Jimenez, J., Canagaratna, M., Donahue, N., Prevot, A., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., and Ng, N.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Jo, D. S., Park, R. J., Lee, S., Kim, S.-W., and Zhang, X.: A global simulation of brown carbon: implications for photochemistry and direct radiative effect, Atmos. Chem. Phys., 16, 3413–3432, https://doi.org/10.5194/acp-16-3413-2016, 2016.
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005.
Kang, E., Root, M. J., Toohey, D. W., and Brune, W. H.: Introducing the concept of Potential Aerosol Mass (PAM), Atmos. Chem. Phys., 7, 5727–5744, https://doi.org/10.5194/acp-7-5727-2007, 2007.
Kim, H., Liu, S., Russell, L. M., and Paulson, S. E.: Dependence of real refractive indices on O : C, H : C and mass fragments of secondary organic aerosol generated from ozonolysis and photooxidation of limonene and α-pinene, Aerosol Sci. Tech., 48, 498–507, https://doi.org/10.1080/02786826.2014.893278, 2014.
Kroll, J. H., Donahue, N. M., Jimenez, J. L., Kessler, S. H., Canagaratna, M. R., Wilson, K. R., Altieri, K. E., Mazzoleni, L. R., Wozniak, A. S., and Bluhm, H.: Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 3, 133, https://doi.org/10.1038/NCHEM.948, 2011.
Lack, D. A., Langridge, J. M., Bahreini, R., Cappa, C. D., Middlebrook, A. M., and Schwarz, J. P.: Brown carbon and internal mixing in biomass burning particles, Proc. Natl. Acad. Sci. USA, 109, 14802–14807, https://doi.org/10.1073/pnas.1206575109, 2012.
Lambe, A. T., Onasch, T. B., Massoli, P., Croasdale, D. R., Wright, J. P., Ahern, A. T., Williams, L. R., Worsnop, D. R., Brune, W. H., and Davidovits, P.: Laboratory studies of the chemical composition and cloud condensation nuclei (CCN) activity of secondary organic aerosol (SOA) and oxidized primary organic aerosol (OPOA), Atmos. Chem. Phys., 11, 8913–8928, https://doi.org/10.5194/acp-11-8913-2011, 2011.
Lambe, A., Massoli, P., Zhang, X., Canagaratna, M., Nowak, J., Daube, C., Yan, C., Nie, W., Onasch, T., Jayne, J., Kolb, C., Davidovits, P., Worsnop, D., and Brune, W.: Controlled nitric oxide production via O(1D) + N2O reactions for use in oxidation flow reactor studies, Atmos. Meas. Tech., 10, 2283–2298, https://doi.org/10.5194/amt-10-2283-2017, 2017.
Laskin, A., Smith, J. S., and Laskin, J.: Molecular characterization of nitrogen-containing organic compounds in biomass burning aerosols using high-resolution mass spectrometry, Environ. Sci. Technol., 43, 3764–3771, https://doi.org/10.1021/es803456n, 2009.
Laskin, A., Laskin, J., and Nizkorodov, S. A.: Chemistry of atmospheric brown carbon, Chem. Rev., 115, 4335–4382, https://doi.org/10.1021/cr5006167, 2015.
Lee, H. J., Aiona, P. K., Laskin, A., Laskin, J., and Nizkorodov, S. A.: Effect of solar radiation on the optical properties and molecular composition of laboratory proxies of atmospheric brown carbon, Environ. Sci. Technol., 48, 10217–10226, https://doi.org/10.1021/es502515r, 2014.
Levinson, R., Akbari, H., and Berdahl, P.: Measuring solar reflectance-Part I: Defining a metric that accurately predicts solar heat gain, Sol. Energy, 84, 1717–1744, https://doi.org/10.1016/j.solener.2010.04.018, 2010.
Li, C., Ma, Z., Chen, J., Wang, X., Ye, X., Wang, L., Yang, X., Kan, H., Donaldson, D., and Mellouki, A.: Evolution of biomass burning smoke particles in the dark, Atmos. Environ., 120, 244–252, https://doi.org/10.1016/j.atmosenv.2015.09.003, 2015.
Li, C., Hu, Y., Zhang, F., Chen, J., Ma, Z., Ye, X., Yang, X., Wang, L., Tang, X., Zhang, R., Mu, M., Wang, G., Kan, H., Wang, X., and Mellouki, A.: Multi-pollutant emissions from the burning of major agricultural residues in China and the related health-economic effects, Atmos. Chem. Phys., 17, 4957–4988, https://doi.org/10.5194/acp-17-4957-2017, 2017.
Li, Y. J., Yeung, J. W., Leung, T. P., Lau, A. P., and Chan, C. K.: Characterization of organic particles from incense burning using an aerodyne high-resolution time-of-flight aerosol mass spectrometer, Aerosol Sci. Tech., 46, 654–665, https://doi.org/10.1080/02786826.2011.653017, 2012.
Li, Y. J., Huang, D. D., Cheung, H. Y., Lee, A. K. Y., and Chan, C. K.: Aqueous-phase photochemical oxidation and direct photolysis of vanillin – a model compound of methoxy phenols from biomass burning, Atmos. Chem. Phys., 14, 2871–2885, https://doi.org/10.5194/acp-14-2871-2014, 2014.
Lin, P., Liu, J., Shilling, J. E., Kathmann, S. M., Laskin, J., and Laskin, A.: Molecular characterization of brown carbon (BrC) chromophores in secondary organic aerosol generated from photo-oxidation of toluene, Phys. Chem. Chem. Phys., 17, 23312–23325, https://doi.org/10.1039/C5CP02563J, 2015.
Lin, P., Aiona, P. K., Li, Y., Shiraiwa, M., Laskin, J., Nizkorodov, S. A., and Laskin, A.: Molecular characterization of brown carbon in biomass burning aerosol particles, Environ. Sci. Technol., 50, 11815–11824, https://doi.org/10.1021/acs.est.6b03024, 2016.
Lin, P., Bluvshtein, N., Rudich, Y., Nizkorodov, S. A., Laskin, J., and Laskin, A.: Molecular Chemistry of Atmospheric Brown Carbon Inferred from a Nationwide Biomass Burning Event, Environ. Sci. Technol., 51, 11561–11570, https://doi.org/10.1021/acs.est.7b02276, 2017.
Lin, P., Fleming, L. T., Nizkorodov, S. A., Laskin, J., and Laskin, A.: Comprehensive Molecular Characterization of Atmospheric Brown Carbon by High Resolution Mass Spectrometry with Electrospray and Atmospheric Pressure Photoionization, Anal. Chem., 90, 12493–12502, https://doi.org/10.1021/acs.analchem.8b02177, 2018.
Liu, J., Bergin, M., Guo, H., King, L., Kotra, N., Edgerton, E., and Weber, R. J.: Size-resolved measurements of brown carbon in water and methanol extracts and estimates of their contribution to ambient fine-particle light absorption, Atmos. Chem. Phys., 13, 12389–12404, https://doi.org/10.5194/acp-13-12389-2013, 2013.
Liu, J., Lin, P., Laskin, A., Laskin, J., Kathmann, S. M., Wise, M., Caylor, R., Imholt, F., Selimovic, V., and Shilling, J. E.: Optical properties and aging of light-absorbing secondary organic aerosol, Atmos. Chem. Phys., 16, 12815–12827, https://doi.org/10.5194/acp-16-12815-2016, 2016.
Lu, J. W., Flores, J. M., Lavi, A., Abo-Riziq, A., and Rudich, Y.: Changes in the optical properties of benzo[a] pyrene-coated aerosols upon heterogeneous reactions with NO2 and NO3, Phys. Chem. Chem. Phys., 13, 6484–6492, https://doi.org/10.1039/C0CP02114H, 2011.
McDonald, J. D., Zielinska, B., Fujita, E. M., Sagebiel, J. C., Chow, J. C., and Watson, J. G.: Fine particle and gaseous emission rates from residential wood combustion, Environ. Sci. Technol., 34, 2080–2091, https://doi.org/10.1021/es9909632, 2000.
Miljevic, B., Hedayat, F., Stevanovic, S., Fairfull-Smith, K. E., Bottle, S., and Ristovski, Z.: To sonicate or not to sonicate PM filters: reactive oxygen species generation upon ultrasonic irradiation, Aerosol Sci. Tech., 48, 1276–1284, https://doi.org/10.1080/02786826.2014.981330, 2014.
Moise, T., Flores, J. M., and Rudich, Y.: Optical properties of secondary organic aerosols and their changes by chemical processes, Chem. Rev., 115, 4400–4439, https://doi.org/10.1021/cr5005259, 2015.
Mutzel, A., Rodigast, M., Iinuma, Y., Böge, O., and Herrmann, H.: An improved method for the quantification of SOA bound peroxides, Atmos. Environ., 67, 365–369, https://doi.org/10.1016/j.atmosenv.2012.11.012, 2013.
Ng, N. L., Chhabra, P. S., Chan, A. W. H., Surratt, J. D., Kroll, J. H., Kwan, A. J., McCabe, D. C., Wennberg, P. O., Sorooshian, A., Murphy, S. M., Dalleska, N. F., Flagan, R. C., and Seinfeld, J. H.: Effect of NOx level on secondary organic aerosol (SOA) formation from the photooxidation of terpenes, Atmos. Chem. Phys., 7, 5159–5174, https://doi.org/10.5194/acp-7-5159-2007, 2007.
Ng, N. L., Canagaratna, M. R., Zhang, Q., Jimenez, J. L., Tian, J., Ulbrich, I. M., Kroll, J. H., Docherty, K. S., Chhabra, P. S., Bahreini, R., Murphy, S. M., Seinfeld, J. H., Hildebrandt, L., Donahue, N. M., DeCarlo, P. F., Lanz, V. A., Prévôt, A. S. H., Dinar, E., Rudich, Y., and Worsnop, D. R.: Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry, Atmos. Chem. Phys., 10, 4625–4641, https://doi.org/10.5194/acp-10-4625-2010, 2010.
Ng, N. L., Canagaratna, M. R., Jimenez, J. L., Chhabra, P. S., Seinfeld, J. H., and Worsnop, D. R.: Changes in organic aerosol composition with aging inferred from aerosol mass spectra, Atmos. Chem. Phys., 11, 6465–6474, https://doi.org/10.5194/acp-11-6465-2011, 2011.
Park, R. J., Kim, M. J., Jeong, J. I., Youn, D., and Kim, S.: A contribution of brown carbon aerosol to the aerosol light absorption and its radiative forcing in East Asia, Atmos. Environ., 44, 1414–1421, https://doi.org/10.1016/j.atmosenv.2010.01.042, 2010.
Passig, J., Schade, J., Oster, M., Fuchs, M., Ehlert, S., Jäger, C., Sklorz, M., and Zimmermann, R.: Aerosol mass spectrometer for simultaneous detection of polyaromatic hydrocarbons and inorganic components from individual particles, Anal. Chem., 89, 6341–6345, https://doi.org/10.1021/acs.analchem.7b01207, 2017.
Peng, Z. and Jimenez, J. L.: Modeling of the chemistry in oxidation flow reactors with high initial NO, Atmos. Chem. Phys., 17, 11991–12010, https://doi.org/10.5194/acp-17-11991-2017, 2017.
Peng, Z., Day, D. A., Stark, H., Li, R., Lee-Taylor, J., Palm, B. B., Brune, W. H., and Jimenez, J. L.: HOx radical chemistry in oxidation flow reactors with low-pressure mercury lamps systematically examined by modeling, Atmos. Meas. Tech., 8, 4863–4890, https://doi.org/10.5194/amt-8-4863-2015, 2015.
Peng, Z., Day, D. A., Ortega, A. M., Palm, B. B., Hu, W., Stark, H., Li, R., Tsigaridis, K., Brune, W. H., and Jimenez, J. L.: Non-OH chemistry in oxidation flow reactors for the study of atmospheric chemistry systematically examined by modeling, Atmos. Chem. Phys., 16, 4283–4305, https://doi.org/10.5194/acp-16-4283-2016, 2016.
Pettersson, A., Lovejoy, E. R., Brock, C. A., Brown, S. S., and Ravishankara, A.: Measurement of aerosol optical extinction at 532 nm with pulsed cavity ring down spectroscopy, J. Aerosol Sci., 35, 995–1011, https://doi.org/10.1016/j.jaerosci.2004.02.008, 2004.
Phillips, S. M. and Smith, G. D.: Light absorption by charge transfer complexes in brown carbon aerosols, Environ. Sci. Technol. Lett., 1, 382–386, https://doi.org/10.1021/ez500263j, 2014.
Pósfai, M., Gelencsér, A., Simonics, R., Arató, K., Li, J., Hobbs, P. V., and Buseck, P. R.: Atmospheric tar balls: Particles from biomass and biofuel burning, J. Geophys. Res.-Atmos., 109, D06213, https://doi.org/10.1029/2003JD004169, 2004.
Rajput, P. and Sarin, M.: Polar and non-polar organic aerosols from large-scale agricultural-waste burning emissions in Northern India: implications to organic mass-to-organic carbon ratio, Chemosphere, 103, 74–79, https://doi.org/10.1016/j.chemosphere.2013.11.028, 2014.
Reid, J. S., Eck, T. F., Christopher, S. A., Koppmann, R., Dubovik, O., Eleuterio, D. P., Holben, B. N., Reid, E. A., and Zhang, J.: A review of biomass burning emissions part III: intensive optical properties of biomass burning particles, Atmos. Chem. Phys., 5, 827–849, https://doi.org/10.5194/acp-5-827-2005, 2005.
Rettner, C. T. and Brophy, J. H.: Resonance enhanced laser ionisation mass spectrometry of four aromatic molecules, Chem. Phys., 56, 53–61, https://doi.org/10.1016/0301-0104(81)85099-9, 1981.
Rollins, A. W., Browne, E. C., Min, K. E., Pusede, S. E., Wooldridge, P. J., Gentner, D. R., Goldstein, A. H., Liu, S., Day, D. A., and Russell, L. M.: Evidence for NOx control over nighttime SOA formation, Science, 337, 1210–1212, https://doi.org/10.1126/science.1221520, 2012.
Rudich, Y.: Data from: Dynamic changes of optical and chemical properties of tar ball aerosols by atmospheric photochemical aging [electronic resource], Weizmann Institute of Science, available at: https://weizmann.alma.exlibrisgroup.com/view/delivery/972WIS_INST/1245276980003596, last access: 25 December 2018.
Rudich, Y., Donahue, N. M., and Mentel, T. F.: Aging of organic aerosol: Bridging the gap between laboratory and field studies, Annu. Rev. Phys. Chem., 58, 321–352, https://doi.org/10.1146/annurev.physchem.58.032806.104432, 2007.
Russell, P. B., Bergstrom, R. W., Shinozuka, Y., Clarke, A. D., DeCarlo, P. F., Jimenez, J. L., Livingston, J. M., Redemann, J., Dubovik, O., and Strawa, A.: Absorption Angstrom Exponent in AERONET and related data as an indicator of aerosol composition, Atmos. Chem. Phys., 10, 1155–1169, https://doi.org/10.5194/acp-10-1155-2010, 2010.
Samburova, V., Connolly, J., Gyawali, M., Yatavelli, R. L., Watts, A. C., Chakrabarty, R. K., Zielinska, B., Moosmüller, H., and Khlystov, A.: Polycyclic aromatic hydrocarbons in biomass-burning emissions and their contribution to light absorption and aerosol toxicity, Sci. Total Environ., 568, 391–401, https://doi.org/10.1016/j.scitotenv.2016.06.026, 2016.
Schauer, J. J., Kleeman, M. J., Cass, G. R., and Simoneit, B. R.: Measurement of emissions from air pollution sources. 3. C1-C29 organic compounds from fireplace combustion of wood, Environ. Sci. Technol., 35, 1716–1728, https://doi.org/10.1021/es001331e, 2001.
Sedlacek III, A. J., Buseck, P. R., Adachi, K., Onasch, T. B., Springston, S. R., and Kleinman, L.: Formation and evolution of tar balls from northwestern US wildfires, Atmos. Chem. Phys., 18, 11289–11301, https://doi.org/10.5194/acp-18-11289-2018, 2018.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: from air pollution to climate change, 3rd edition, John Wiley & Sons, Inc., Hoboken, New Jersey, USA, 2016.
Sengupta, D., Samburova, V., Bhattarai, C., Kirillova, E., Mazzoleni, L., Iaukea-Lum, M., Watts, A., Moosmüller, H., and Khlystov, A.: Light absorption by polar and non-polar aerosol compounds from laboratory biomass combustion, Atmos. Chem. Phys., 18, 10849–10867, https://doi.org/10.5194/acp-18-10849-2018, 2018.
Shamjad, P. M., Satish, R. V., Thamban, N. M., Rastogi, N., and Tripathi, S.: Absorbing refractive index and direct radiative forcing of atmospheric Brown Carbon over Gangetic Plain, ACS Earth Space Chem., 2, 31–37, https://doi.org/10.1021/acsearthspacechem.7b00074, 2018.
Shen, G., Tao, S., Wei, S., Zhang, Y., Wang, R., Wang, B., Li, W., Shen, H., Huang, Y., and Yang, Y.: Retene emission from residential solid fuels in China and evaluation of retene as a unique marker for soft wood combustion, Environ. Sci. Technol., 46, 4666–4672, https://doi.org/10.1021/es300144m, 2012.
Shen, H., Huang, Y., Wang, R., Zhu, D., Li, W., Shen, G., Wang, B., Zhang, Y., Chen, Y., and Lu, Y.: Global atmospheric emissions of polycyclic aromatic hydrocarbons from 1960 to 2008 and future predictions, Environ. Sci. Technol., 47, 6415–6424, https://doi.org/10.1021/es400857z, 2013.
Shrivastava, M., Cappa, C. D., Fan, J., Goldstein, A. H., Guenther, A. B., Jimenez, J. L., Kuang, C., Laskin, A., Martin, S. T., and Ng, N. L.: Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 55, 509–559, https://doi.org/10.1002/2016RG000540, 2017.
Sigsgaard, T., Forsberg, B., Annesi-Maesano, I., Blomberg, A., Bølling, A., Boman, C., Bønløkke, J., Brauer, M., Bruce, N., and Héroux, M. E.: Health impacts of anthropogenic biomass burning in the developed world, Eur. Respir. J., 46, 1577–1588, https://doi.org/10.1183/13993003.01865-2014, 2015.
Sumlin, B. J., Pandey, A., Walker, M. J., Pattison, R. S., Williams, B. J., and Chakrabarty, R. K.: Atmospheric Photooxidation Diminishes Light Absorption by Primary Brown Carbon Aerosol from Biomass Burning, Environ. Sci. Technol. Lett., 4, 540–545, https://doi.org/10.1021/acs.estlett.7b00393, 2017.
Sumlin, B. J., Oxford, C. R., Seo, B., Pattison, R. R., Williams, B. J., and Chakrabarty, R. K.: Density and homogeneous internal composition of primary brown carbon aerosol, Environ. Sci. Technol., 52, 3982–3989, https://doi.org/10.1021/acs.est.8b00093, 2018.
Takahama, S., Johnson, A., Morales, J. G., Russell, L. M., Duran, R., Rodriguez, G., Zheng, J., Zhang, R., Toom-Sauntry, D., and Leaitch, W. R.: Submicron organic aerosol in Tijuana, Mexico, from local and Southern California sources during the CalMex campaign, Atmos. Environ., 70, 500–512, https://doi.org/10.1016/j.atmosenv.2012.07.057, 2013.
Tavakoli, F. and Olfert, J.: An instrument for the classification of aerosols by particle relaxation time: theoretical models of the aerodynamic aerosol classifier, Aerosol Sci. Tech., 47, 916–926, https://doi.org/10.1080/02786826.2013.802761, 2013.
Tavakoli, F. and Olfert, J. S.: Determination of particle mass, effective density, mass-mobility exponent, and dynamic shape factor using an aerodynamic aerosol classifier and a differential mobility analyzer in tandem, J. Aerosol Sci., 75, 35–42, https://doi.org/10.1016/j.jaerosci.2014.04.010, 2014.
Tóth, A., Hoffer, A., Nyiro-Kósa, I., Pósfai, M., and Gelencsér, A.: Atmospheric tar balls: aged primary droplets from biomass burning?, Atmos. Chem. Phys., 14, 6669–6675, https://doi.org/10.5194/acp-14-6669-2014, 2014.
Washenfelder, R. A., Flores, J. M., Brock, C. A., Brown, S. S., and Rudich, Y.: Broadband measurements of aerosol extinction in the ultraviolet spectral region, Atmos. Meas. Tech., 6, 861–877, https://doi.org/10.5194/amt-6-861-2013, 2013.
Washenfelder, R., Attwood, A., Brock, C., Guo, H., Xu, L., Weber, R., Ng, N., Allen, H., Ayres, B., and Baumann, K.: Biomass burning dominates brown carbon absorption in the rural southeastern United States, Geophys. Res. Lett., 42, 653–664, https://doi.org/10.1002/2014GL062444, 2015.
Wei, C., Bandowe, B. A. M., Han, Y., Cao, J., Zhan, C., and Wilcke, W.: Polycyclic aromatic hydrocarbons (PAHs) and their derivatives (alkyl-PAHs, oxygenated-PAHs, nitrated-PAHs and azaarenes) in urban road dusts from Xi'an, Central China, Chemosphere, 134, 512–520, https://doi.org/10.1016/j.chemosphere.2014.11.052, 2015.
Weimer, S., Alfarra, M., Schreiber, D., Mohr, M., Prévôt, A., and Baltensperger, U.: Organic aerosol mass spectral signatures from wood-burning emissions: Influence of burning conditions and wood type, J. Geophys. Res.-Atmos., 113, D10304, https://doi.org/10.1029/2007JD009309, 2008.
Wong, J. P., Zhou, S., and Abbatt, J. P.: Changes in secondary organic aerosol composition and mass due to photolysis: relative humidity dependence, J. Phys. Chem. A, 119, 4309–4316, https://doi.org/10.1021/jp506898c, 2014.
Xie, M., Hays, M. D., and Holder, A. L.: Light-absorbing organic carbon from prescribed and laboratory biomass burning and gasoline vehicle emissions, Sci. Rep. UK, 7, 7318, https://doi.org/10.1038/s41598-017-06981-8, 2017.
Yee, L. D., Kautzman, K. E., Loza, C. L., Schilling, K. A., Coggon, M. M., Chhabra, P. S., Chan, M. N., Chan, A. W. H., Hersey, S. P., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from biomass burning intermediates: phenol and methoxyphenols, Atmos. Chem. Phys., 13, 8019–8043, https://doi.org/10.5194/acp-13-8019-2013, 2013.
Yu, L., Smith, J., Laskin, A., Anastasio, C., Laskin, J., and Zhang, Q.: Chemical characterization of SOA formed from aqueous-phase reactions of phenols with the triplet excited state of carbonyl and hydroxyl radical, Atmos. Chem. Phys., 14, 13801–13816, https://doi.org/10.5194/acp-14-13801-2014, 2014.
Zhang, H. and Ying, Q.: Secondary organic aerosol from polycyclic aromatic hydrocarbons in Southeast Texas, Atmos. Environ., 55, 279–287, https://doi.org/10.1016/j.atmosenv.2012.03.043, 2012.
Zhang, X. and Seinfeld, J. H.: A functional group oxidation model (FGOM) for SOA formation and aging, Atmos. Chem. Phys., 13, 5907–5926, https://doi.org/10.5194/acp-13-5907-2013, 2013.
Zhong, M. and Jang, M.: Dynamic light absorption of biomass-burning organic carbon photochemically aged under natural sunlight, Atmos. Chem. Phys., 14, 1517–1525, https://doi.org/10.5194/acp-14-1517-2014, 2014.
Zhou, S., Collier, S., Jaffe, D. A., Briggs, N. L., Hee, J., Sedlacek III, A. J., Kleinman, L., Onasch, T. B., and Zhang, Q.: Regional influence of wildfires on aerosol chemistry in the western US and insights into atmospheric aging of biomass burning organic aerosol, Atmos. Chem. Phys., 17, 2477–2493, https://doi.org/10.5194/acp-17-2477-2017, 2017.