the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 08 Nov 2019
| 08 Nov 2019
Secondary organic aerosol formation from photooxidation of furan: effects of NOx and humidity
Xiaotong Jiang
Narcisse T. Tsona
Long Jia
Shijie Liu
Hailiang Zhang
Yongfu Xu
Atmospheric furan is a primary and secondary pollutant in the atmosphere, and its emission contributes to the formation of ultrafine particles. We investigate the effects of NOx level and humidity on the formation of secondary organic aerosol (SOA) generated from the photooxidation of furan in the presence of NaCl seed particles. SOA mass concentration and yield were determined under different NOx and humidity levels. A significant difference is observed both in the variation of SOA mass concentration and SOA yield with the initial experimental conditions. Varying VOC (volatile organic compound) ∕ NOx ratios over the range 48.1 to 8.2 contributes to the effective formation of SOA in the presence of NaCl seed particles, with the SOA mass concentration and SOA yield ranging from 0.96 to 23.46 µg m−3 and from 0.04 % to 1.01 %, respectively. We found that there was a favourable relationship between the SOA yields and NOx concentration. In particular, the increase in SOA yield with increasing NOx concentration was continuously observed at high NOx levels owing to a corresponding increase in the amount of low-volatility hydroxyl nitrates and dihydroxyl dinitrates that can partition into the particle phase. In addition, varying relative humidity (RH) from 5 % to 88 % increased the SOA yield from 1.01 % to 5.03 %. The enhanced SOA formation from humid conditions may result from the high OH concentration, rapid furan decay rate, enhanced carbonyl-rich products condensation, and the aqueous-phase reactions. Using hybrid quadrupole-orbitrap mass spectrometer equipped with electrospray ionization (HESI-Q Exactive-Orbitrap MS), three carbonyl-rich products and three kinds of organonitrates were identified in the collected SOA. Based on the HESI-Q Exactive-Orbitrap MS analysis and Fourier transform infrared spectroscopy (FTIR), the reaction mechanism of furan photooxidation was proposed. This study demonstrates the effects of NOx and humidity on SOA formation during the furan–NOx–NaCl photooxidation and provides new insights into the oxidation regime and SOA composition in furan photooxidation. The results also illustrate the importance of studying SOA formation over a comprehensive range of environmental conditions. Only such evaluations can induce meaningful SOA mechanisms to be implemented in air quality models.
- Article
(4065 KB) -
Supplement
(1386 KB) - BibTeX
- EndNote





Atmospheric particulate matter (PM) is primarily composed of organic carbon, elemental carbon, sulfate, nitrate, and other components (Donahue et al., 2009; Zhang et al., 2011), which have adverse effects on human health and global climate forcing (Hallquist et al., 2009; Pope III et al., 2013). Secondary organic aerosols (SOAs) constitute a substantial portion of the total ambient aerosol particles, which mainly originate from biomass burning and atmospheric reactions of volatile organic compounds (VOCs) (Kanakidou et al., 2005). Identifying the chemical composition and major precursors of SOA is helpful to better understand their formation mechanism and strategies for the control of PM. Furan is an important five-member heteroatom-containing VOC present in the atmosphere, and it can be produced by wood combustion, oil refining, coal mining and gasification, and pyrolysis of biomass, cellulose, and lignin (Shafizadeh, 1982). As shown in previous studies, furan is one of the most common products of the thermal cracking of biomass (Kahan et al., 2013; Gilman et al., 2015). Both furan and furan-related compounds have been detected in the effluent from initial smouldering of fuels, oats, and soft pellets (Olsson, 2006; Perzon, 2010). In addition, furan has been identified as one of the major compounds emitted from the combustion of ponderosa pine, which constitute a significant fraction (5 %–37 % by estimated emission factor) of smoke from combustion (Hatch et al., 2015). Field measurements of hydrocarbon emissions from biomass burning in Brazil have also shown that furans consist of 52 % and 72 % of the oxygenated hydrocarbons emissions in the Cerrado (grasslands) and selva (tropical forest) regions, respectively (Greenberg et al., 1984). Moreover, furan has been proven to be the typical trace gas of roasting or burning activities (Gloess et al., 2014; Coggon et al., 2016; Burling et al., 2010; Gilman et al., 2015; Sarkar et al., 2016; Stockwell et al., 2016, 2015, 2014), and it is also an important contribution to OH reactivity towards biomass burning emissions (Gilman et al., 2015; Sarkar et al., 2016). Furan is also a secondary pollutant produced in the photooxidation of some conjugate alkenes. Previous product studies of 1,3-butadiene oxidation have shown that furan is one of the dominant product during the OH radical-initiated reactions in the presence of NO (Sprengnether et al., 2002; Tuazon et al., 1999). Furthermore, another source of furan is the cyclization of the unsaturated 1,4-hydroxyaldehydes, which will further go through the dehydration reactions (Baker et al., 2005). There is strong evidence that furan can cause malignant tumour formation in a variety of experimental animals (IARC, 1995). Therefore, a better understanding of the atmospheric transformations of furan will be helpful to improve the air quality models for toxic species.
As an aromatic hydrocarbon, furan is very reactive according to its electrophilic substitution reactions (Villanueva et al., 2007). The measured rate constant of furan with OH radicals, O3, and NO3 radicals at room temperature is , , and cm3 molecule−1 s−1, respectively (Atkinson et al., 1983, 1985). If the oxidant concentration of OH and NO3 is assumed to be 1.6×106 and 5×108 molecules cm−3, lifetimes of furan for reactions with OH and NO3 are estimated to be 3 h and 18 min, respectively (Cabanas et al., 2004). In the troposphere, atmospheric furan is expected to be mainly oxidized by OH during the daytime and NO3 at night. It is important to assess the SOA formation potential of furan and its role in SOA production in biomass burning plumes. Although the SOA formation potential of furan has been studied in recent years (Cabanas et al., 2004; Liljegren and Stevens, 2013; Tapia et al., 2011), the influence of several factors including NOx levels and relative humidity (RH) on SOA formation should be further investigated. It is generally accepted that NOx level plays a critical role in SOA formation, by governing the reactions of organo-peroxy radicals (RO2) (Song et al., 2007, 2005). The branching ratio of RO2 reactions with NOx and the hydroperoxyl radical (HO2) is determined by the NOx level during experiments (Kroll and Seinfeld, 2008). A previous study showed that the mechanisms of the two reactions differ sharply under different NOx level conditions. RO2 reacts only with NO under high NOx levels and with HO2 under low-NOx conditions (Ng et al., 2007a). The latter case produces lower-volatility products than the former one. An increase in SOA yield with increasing NOx was proposed to be due to the formation of low-volatility compounds including multifunctional nitrates and dinitrates, which partitioned to the particle phase and contribute significantly to isoprene SOA under high-NOx conditions (Schwantes et al., 2019). Another study of the SOA formation from the irradiation of propylene also showed that the SOA yield decreased with increasing propylene ∕ NOx ratio (Ge et al., 2017a). Recently, C. Liu et al. (2019) indicated that NO2 could participate in the OH-initiated reaction of guaiacol, consequently resulting in the formation of organic nitrates and promoting SOA formation (C. Liu et al., 2019). The effect of NOx level on SOA formation from aromatic precursors has been investigated previously, but the results are inconclusive. A study focusing on the photooxidation of toluene and m-xylene has demonstrated that aerosol yields decrease as NOx level increases (Xu et al., 2015), while another study showed that the SOA yield from photooxidation of isoprene under high NO2∕NO is 3 times more important than that measured under low NO2∕NO (Chan et al., 2010).
Besides the mixed effect of NOx level on SOA formation, the RH can significantly alter the sizes of SOA particles, depending on their hygroscopicity (Varutbangkul et al., 2006), their gas-phase reactions involving water (Jonsson et al., 2006), and the aqueous chemistry occurring at their surfaces (Lim et al., 2010; Grgic et al., 2010). The photooxidation experiments with aromatic compounds, such as toluene (Edney et al., 2000; Cao and Jang, 2010; Faust et al., 2017; Hinks et al., 2018), benzene and ethylbenzene (Jia and Xu, 2014), m-xylene (Zhou et al., 2011), and 1,3,5-trimethylbenzene (Cocker et al., 2001) have been carried out previously to study the effect of RH on SOA formation. The results exhibited large discrepancies under different experimental conditions. The yield of SOA generated under low-NOx conditions was found to be small at high RH (Cao and Jang, 2010). A little RH effect was also observed on SOA particle formation and size distributions (Bonn and Moortgat, 2002; Fry et al., 2009). Nevertheless, positive correlations between RH and SOA yield have also been observed in the presence of hygroscopic seed particles (Zhou et al., 2011), and the role of RH in SOA formation also appears to be mixed. Water vapour not only participates in the VOC photooxidation reactions and affects the gas-phase oxidation mechanisms but also alters the partitioning of the reaction products between the gas phase and the particle phase, thereby influencing the equilibrium partitioning of generated organic aerosols (Spittler et al., 2006). Moreover, the RH is connected with SOA formation due to its strong influence on seed acidity and particle liquid water concentration (Mahowald et al., 2011). A high-RH environment contributes to the increase in aerosol liquid water (ALW), which promotes the hydrolysis of organic compounds and leads to other heterogeneous and aqueous-phase reactions (Ervens et al., 2011). Previous studies regarding the atmospheric reactions of furan typically focused on the kinetics and mechanism (Gomez Alvarez et al., 2009; Aschmann et al., 2014).
In the present study, we used FTIR, in concert with a scanning mobility particle sizer (SMPS) to elucidate the roles of NOx level and RH in SOA formation from furan–NOx irradiation. All the experiments were conducted in the presence of NaCl seed particles, which acted as the nuclei and provided sufficient seed surface area at the beginning of the reaction to suppress the effects of vapour wall losses of semi- or low-volatility species. Specifically, we evaluate whether the increased ALW affects the SOA mass concentration and the SOA yield. The chemical composition of furan SOA was investigated by heated electrospray ionization high-resolution orbitrap mass spectrometer (HESI-Q Exactive-Orbitrap MS), with a focus on the formation of organic nitrates. Strong evidence that both the RH and different NOx levels have a significant effect on SOA formation from furan photooxidation are presented.
2.1 SOA sample preparation
All experiments were performed in a 1.3 m3 Teflon film chamber at the Institute of Atmospheric Physics, Chinese Academy of Sciences, Beijing. To maximize and homogenize the interior light intensity, a mirror surface stainless steel was chosen as the interior wall of the enclosure. A total of 42 black lamps (F40BLB, GE, Fairfield, CT, USA) with an emission band centre at 365 nm were equipped in the facilities to simulate the spectrum of the UV band in solar irradiation. The chamber was equipped with multiple sampling ports, which allowed the introduction of clean air, seed aerosols, and gas-phase reagents and for measurements of both gas-phase and particle-phase compositions. The reaction set-up used in the present study and the schematic of the smog chamber facility are shown in Fig. S1 in the Supplement (Liu et al., 2017; Jia and Xu, 2014, 2016; Ge et al., 2017a, b).
The zero air (generated from Zero Air Supply, Model 111 and Model 1150, Thermo Scientific) was used as the background gas for the experiments, and three big hydrocarbon traps (BHT-4, Agilent, Santa Clara, CA, USA) coupled with three activated-carbon filters were used to get further purified air. The chamber was cleaned by flowing pure air for at least 4 h prior to each experiment until residual NO, NO2, O3, or any other particles could not be detected in the chamber. The prepared gas-phase furan was introduced into the chamber directly by a syringe. A certain amount of NO2 was injected into the chamber by a gas-tight syringe to achieve the initial NOx level. For the enhancement of the SOA formation, NaCl seed aerosols were injected into the chamber via the atomization of NaCl aqueous solution with a constant-rate atomizer (Model 3076, TSI, USA). An aerosol neutralizer (Model 3087, TSI, USA) was used to bring particles to a steady-state charge distribution before they were introduced into the reactor. The initial seed number and mass concentrations were approximately 5×104 cm−3 and 6 µg m−3 on average, respectively. For different RH conditions, the dry zero air was introduced into a bottle of high-purity water to control the humidity of the background air. The humidity in the chamber was detected with a hygrometer (Model 645, Testo AG, Lenzkirch, Germany). The initial experimental conditions considered in this study are summarized in Table 1. Typically, approximate 750 ppb furan was employed. To study the effect of NOx levels on SOA formation, experiments were conducted with initial furan ∕ NOx ratios ranging from 7.8 to 48.1, whereas to develop and test the role of RH, experiments were performed for RH values varying from 5 % to 85 % and with an initial average furan to NOx ratio of 7.55. The temperature of the chamber was controlled to be 308–310 K during all the experiments.
Table 1Summary of initial conditions, O3 concentrations, and particle mass concentrations in furan–NOx–NaCl photooxidation experiments
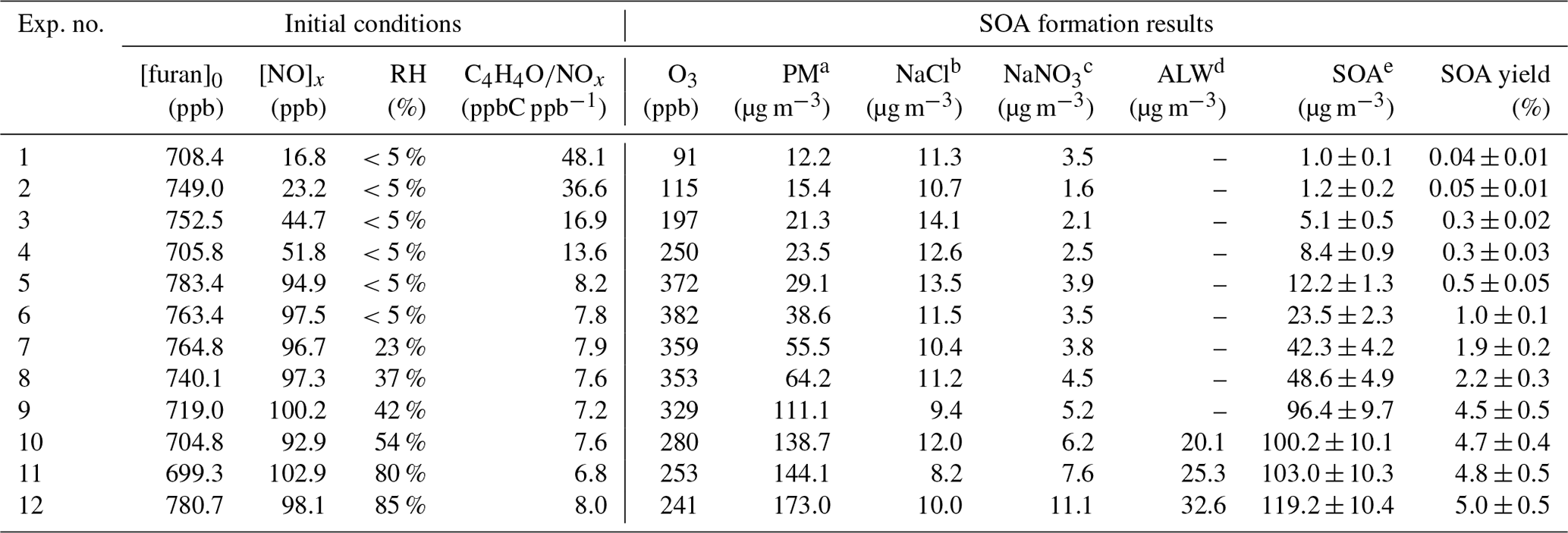
a PM: particle mass concentration in the chamber was determined from the
SMPS and was the sum of NaCl, NaNO3, ALW, and SOA at the end of the
experiments.
b NaCl: the amount of NaCl at the end of the experiments.
c NaNO3: the amount of NaNO3 at the end of the experiments.
d ALW: the amount of aerosol liquid water content
at the end of the
experiments.
e SOA: the amount of secondary organic aerosol at the end of the
experiments.
Particles and gas-phase species may get lost to the chamber walls on short timescales, thereby influencing the gas-phase chemistry and SOA formation. The wall loss rate constants for O3, NOx, and aerosol particles were , , and s−1, respectively, which were detected from our previous study conducted in the same set-up and similar experimental conditions (Ge et al., 2017a). However, no wall loss of furan was observed within the uncertainty of the detection of the instrument. Furthermore, the light intensity in the reactor was determined to be 0.34 min−1 by using the NO2 photolysis rate constant as the indirect representation. Before the start of each experiment, the static electricity of the Teflon chamber was removed. After all the reactants flowed into the chamber, the reactor was maintained in the dark for at least 40 min without any activities to reach homogeneous mixing.
2.2 Gas and particle monitoring
The concentrations of O3 and NOx were measured continuously by a UV photometric O3 analyser (Model 49C, Thermo Environmental Instruments Inc.) and a chemiluminescence analyser (Model 42C, Thermo Electron Corporation, USA), respectively. The sampling flow rate was 0.75 L min−1 for both O3 and NOx analysers. The uncertainty in the gas-phase measurements was below ±1 %. The experiment samples were collected by Tenax absorption tubes (150 mm length × 6 mm O.D., 0.2 g sorbent). Furan concentrations and product samples were detected by thermal-desorption gas chromatography mass spectrometry (TD-GC-MS): the thermal desorber (Master TD, Dani, Italy) was combined with gas chromatography (Model 7890A, Agilent Tech., USA) interfaced to a mass selective detector (5975C, Agilent Tech., USA). The initial reactant concentrations are given in Table 1. The chromatographic analytical method used for the analysis was as follows: the chromatographic column was kept at 338 K for 4 min, then heated to 598 K at a heating rate of 20 K min−1, and held for 5 min.
The particle size distribution and mass concentration were measured with a scanning mobility particle sizer (SMPS, Model 3936, TSI, USA) composed of a TSI 3081 differential mobility analyser (DMA) and a TSI 3776 condensation particle counter (CPC). The sampling and sheath flow rates of the SMPS were 0.3 and 3 L min−1, respectively. Given this flow conditions of the SMPS, particle sizes (in counts cm−3) were recorded in 64 size bins for particles in the 13.6 to 710.5 nm size range. The accuracy of the particle number concentration is ±10 % at cm−3. The method of the reduced dry ambient size spectrometer (DASS) was used to detect the ALW (Engelhart et al., 2011). To get dry particle mass concentrations, a large diameter Nafion dryer (Permapure MD-700-48F-3) and a multi-tube Nafion dryer (Permapure PD-200T-24E-M) were added to the sampling inlet and sheath flow, respectively. This DASS method is generally accepted and widely used in the detection of ALW content, and can remove up to 90 % of the water vapour without losing the organic-bound water. It should be noted that the dissolved water-soluble species would evaporate back into the gas phase during the ALW measurement when the aerosol water is removed. In fact, the repartitioning of water-soluble components between gas and particle phases was not taken into consideration. The SOA concentrations for high-RH conditions were slightly underestimated, but the underestimation is extremely low and can be negligible. The results were compared with the mass concentrations detected in humid mode, in which the humid air in the chamber was introduced into the SMPS with the sheath air set to 10 L min−1. The ALW was calculated as the difference between the particle mass concentrations determined in dry and humid modes (Jia and Xu, 2018). On the basis of the recorded particle volume concentration and assumed particle density, a total suspended particulate (TSP in µg m−3) could be obtained. As shown in Eq. (1), TSP is the sum of seed aerosol (NaCl in this work), NaNO3, particle ALW, and SOA. By dissolving the SOA collected on ZnSe disks into high-purity water, an ion chromatograph (IC, Dionex ICS-900, Thermo Fisher, USA) was then used to analyse the inorganic content (Cl− and ) in SOA. With known initial NaCl seed aerosols and their decay rates, the particle mass concentration of NaCl can be calculated, and based on the detected ALW during experiments, the SOA produced in the photooxidation of furan can be estimated from Eq. (1).
2.3 Chemical characterization of products
The SOA particles were sampled on ungreased ZnSe disk (25 mm in diameter) using a Dekati low-pressure impactor (DLPI, Dekati Ltd., Kangasala, Finland) after the appearance of the O3 maximum concentration. The sampling flow rate of DLPI was 10 L min−1 with particle sizes from 30 nm to 10 µm, classified into 13 stages. According to the particle distribution explored by the SMPS, the SOA particle size was mainly around 150 nm after 3 h of irradiation. The ZnSe disk was placed on stage 3 to reach the maximum collection of the particles. Afterward, the ZnSe disk was put in the FTIR (Nicolet iS10, Thermos Fisher, USA) sample holder, which had been flushed with N2 to eliminate the impact of ambient H2O and CO2 on the determination of the chemical composition of formed SOA particles. The spectra were recorded at 4 cm−1 resolution with 128 scans. The samples collected on the ZnSe disk were then dissolved with high-purity water for analysing inorganic species.
To obtain chemical characterization information, the ultrahigh performance liquid chromatograph (UPLC; Ultimate 3000, Thermo Scientific, USA) HESI-Q Exactive-Orbitrap MS (Q Exactive, Thermo Scientific, USA) was used for the analysis of organic compounds produced from the photooxidation of furan. Methanol (Optima™ LC/MSGrade, Fisher Chemical, USA) was used as the eluent in the UPLC system. The acquired mass spectrum of SOA was in the range of 80–1000 Da. As a “soft ionization”, HESI can provide molecular formulae for thousands of organic aerosol (OA) substances when coupled with a high-resolution mass analyser. The HESI source was used in both positive and negative ion modes using the optimum method for the characterization of organic compounds. A total volume of 300 L was sampled at a flow rate of 0.2 L min−1, and a volume of 20 µL of the extraction was used for the measurement. The addition of acetonitrile allows a lower surface tension of the solution and provides a stable electrospray ionization process (Koch et al., 2005). N2 was used as the sheath and auxiliary gas. The desolvation gas temperature was 320∘, and the gas flow was 200 µL min−1. The capture voltage was set to 3 kV. The Thermo Scientific Xcalibur software (Thermo Fisher Scientific Inc., USA) was used to analyse the data from HRMS.
3.1 Gas phase and SMPS results
Experiments were divided into two main groups, which were set to study the role of (i) NOx and (ii) RH on SOA formation. To keep the variables of each set of conditions consistent, almost identical initial gas-phase conditions were required. The addition of furan and seed aerosol in all the experiments was nearly equal. The first set of experiments (Exp. 1–6, Table 1), designed to investigate the effect of NOx level on SOA formation, used similar RH conditions with different NOx concentrations. To further assess the effect of OH produced during the furan–NOx photooxidation, four experiments (Table S1 in the Supplement, Exp. 13–16) were conducted by injecting H2O2 into the chamber before the experiments started. For Exp. 6–12, different RH levels coupled with similar furan ∕ NOx ratios were monitored to assess the RH impact on SOA formation. To analyse the SOA composition, five additional experiments (Table S2, Exp. 17–21) were carried out to analyse the HESI-Q Exactive-Orbitrap MS, which intended to compare the role of NOx and RH in the SOA formation. The major gas-phase inorganic chemical reactions which occurred during the experiments are presented in Fig. 1. After turning on the light, the photolysis of NO2 produced NO and O, which further reacted with O2 to form O3 in the chamber. As a result, a slight increase in the beginning followed by a decrease in NOx concentrations are observed (Fig. 2). On one hand, the decrease in NOx is caused by the reaction between NO and alkyl RO2, which are formed from the OH-initiated oxidation of furan during the experiment. On the other hand, the oxidation of NO by O3 also contributes to the NOx decreasing trend.
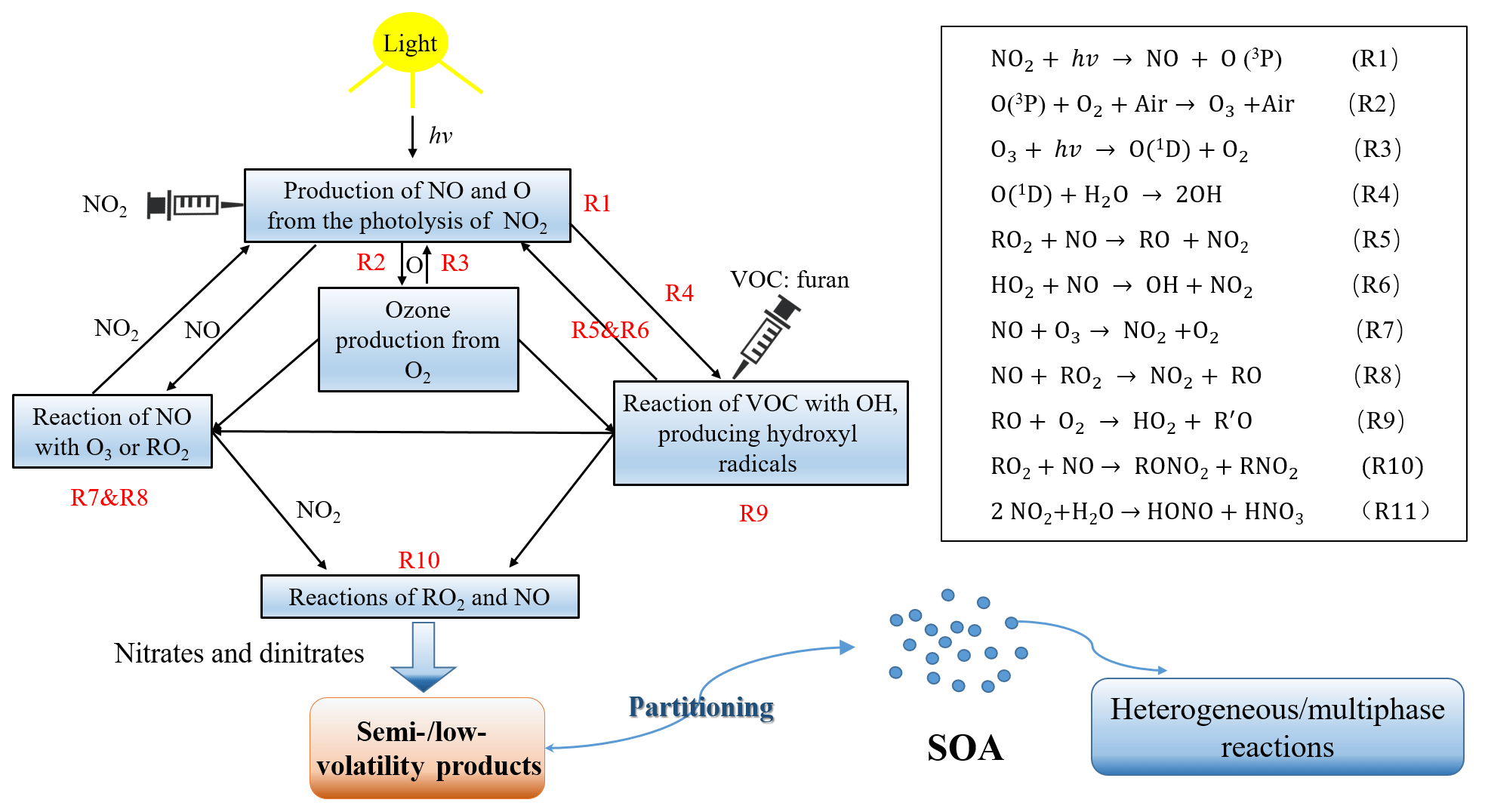
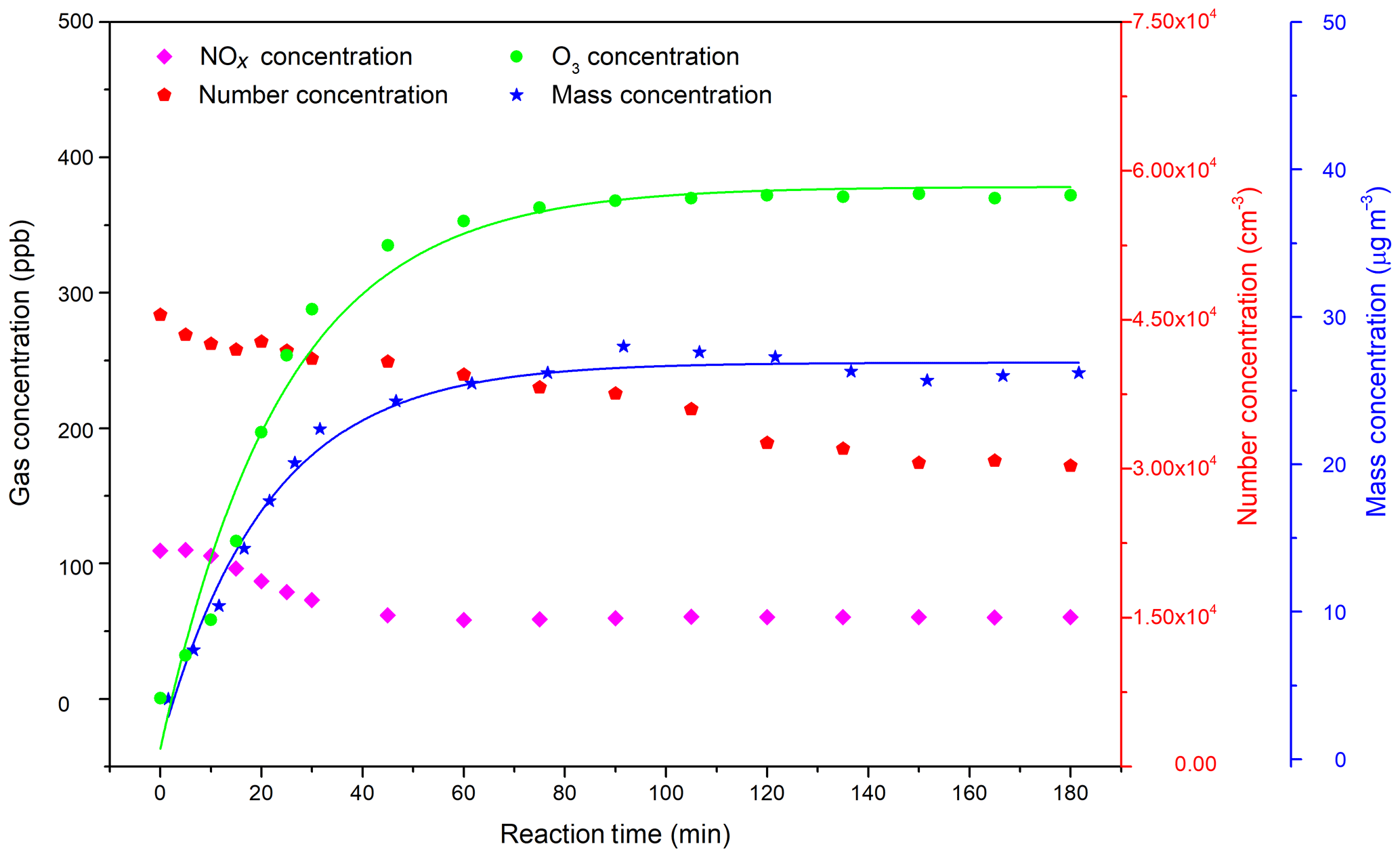
Figure 2Profile of the gas-phase concentrations of reactants (NO, NO2, NOx, and O3) and particle number ∕ mass concentrations (corrected with wall loss) over time. The C4H4O∕NOx ratio is 7.9 and RH=23 %. Since the particle wall loss has a weak RH dependence in our chamber, a mean value of s−1 was used for wall loss correction. A density of 1.4 g cm−3 was used in the SMPS (Jia and Xu, 2018; Kostenidou et al., 2007).
The photochemical oxidation of VOCs in the presence of NOx and OH radical is an important source of tropospheric O3 (Labouze et al., 2004). Although the atmospheric degradation mechanisms of furan are complex, the fundamental features of troposphere O3 formation is relatively simple. The main formation pathway of O3 in the atmosphere is the photolysis of NO2 as presented in Fig. 1 (Reactions R1 and R2). The RO2 and HO2 radicals generated from the photooxidation of furan can further react with NO to form NO2 (Reactions R5 and R6), whose photolysis will produce the ground-state oxygen atom (Reaction R1) and thus contribute to the net O3 formation through the reaction of O2 with O (Reaction R2). The very similar VOC level may result in a high O3 production with higher NOx concentration conditions (Bowman and Seinfeld, 1994). As presented in a previous work on the ethylene–NOx–NaCl irradiations, both experimental results and model simulations showed an abundant O3 production (approximately 500 ppb) with an initial NOx level of 142 ppb (Jia and Xu, 2016). Therefore, it is important to evaluate the O3 production potential of VOCs in the atmosphere. As shown in Fig. 2, it shows a consistent increasing trend between O3 concentration and SOA mass concentration, indicating that O3 was generated during the furan photooxidation reactions with the formation of SOA. However, it should be noted that the apparent high O3 concentration monitored by the O3 analyser was due to species other than O3 that have an absorption around 254 nm. The generated gas-phase reaction products that have carbonyl, carboxylic acid, and ester groups conjugated with C=C bonds can absorb strongly in the ∼ 200–300 nm range and, accordingly, contribute to the apparent intense signal of O3 detected by the O3 monitor (Strollo and Ziemann, 2013).
The O3 maximum concentration, SOA mass concentration and SOA yield dependences on the C4H4O∕NOx ratio are presented in Fig. 2. The results show that the O3 maximum concentration produced during the experiments decreases with increasing C4H4O∕NOx ratio. When the C4H4O∕NOx ratio decreases from 16.9 to 7.8, there is a sharp increase in O3 concentration from 197 to 382 ppb. Interestingly, experiments for which the C4H4O∕NOx ratio changed from 48.1 to 36.60 exhibit a change of 24 ppb in O3 maximum concentration. This result indicates that under different C4H4O∕NOx ratio regimes, the concentration of O3 produced varies much. There is a great change in O3 accumulation when the C4H4O∕NOx ratios are relatively low (<16.9), which is consistent with a previous observation from C3H6–NOx–NaCl irradiation experiments by Ge et al. (2017a), who found that when the C3H6∕NOx ratio was less than 11, the O3 concentration decreased considerably with increasing ratio, whereas when the ratio was larger than 11, the O3 concentration slightly decreased with increasing ratio. Figure S2 shows the comparisons of the observed concentrations of the apparent O3 and NOx from furan irradiations at different experimental conditions. With similar NOx level, the amount of O3 formed at 5 % RH was larger than that at 42 % RH. The apparent O3 maximum concentration was reduced by almost 60 % as the C4H4O∕NOx ratio increased from 7.8 to 36.6. However, the effect of RH on O3 formation is not so obvious compared to the difference in initial C4H4O∕NOx ratio. The influence of NOx level on O3 is likely related to its formation mechanism. The formation of O3 is directly connected with the NOx in two ways: the photolysis of NO2, producing the O atom that reacts with O2, and the NOx reacting directly with RO. However, the RH has little to no effect on O3 formation. This formation is slightly favoured at low RH, whereas, at high RH, the ONO2-containing compounds are easily transferred into the aerosol phase, thereby suppressing the O3 formation (Jia and Xu, 2014). Additionally, the slight change in O3 maximum concentration under different RH conditions may also be caused by the consumption of gas-phase reaction products that contain functional groups conjugated with C=C bonds and respond to the O3 analyser. This assumption is reasonable because these carbonyl-rich products were favourable to condense on the moist surface of particles and thus lowered the O3 concentration detected by the O3 analyser, which has been further confirmed by the MS results with enhanced intensities of corresponding organonitrates. The wall loss of the organonitrates species would also explain the decreasing trend of O3 concentration as RH increases. The appearance time of O3 maximum concentration at a high C4H4O∕NOx ratio is almost half an hour earlier than that at a low initial C4H4O∕NOx ratio. By contrast, there is no significant difference in the balance time of NOx concentration under different experimental conditions. Similar initial C4H4O∕NOx ratios result in a similar profile of NOx concentration during the experiment, although the final NOx concentration shows a slight difference.
The calculated SOA yield is defined as the ratio of the mass concentrations of the maximum SOA formed (MSOA in µg m−3, corrected by wall loss) and the reacted furan (ΔC4H4O in µg m−3):
where MSOA is obtained by subtracting the amount of NaCl and NaNO3 from the total particle mass concentration measured by the SMPS and ΔC4H4O is the consumption of furan during the experiments. Experiments were started with an average of 6 µg m−3 of NaCl seed aerosol and were conducted under different initial C4H4O∕NOx ratios. The dependences of the SOA mass concentration and SOA yield on the C4H4O∕NOx ratios and RH conditions are shown in Figs. 3 and 4, respectively. The numerical values of the aerosol mass concentration and SOA yield are given in Table 1. After the photooxidation reactions, the SOA mass concentrations reached maxima between 1.0 and 23.5 µg m−3, with SOA yield ranging from 0.04 % to 1.01 %. In other experiments carried out under varying RH conditions, SOA maxima were in the range of 23.5–119.2 µg m−3, with SOA yield ranging from 1.01 % to 5.03 %.
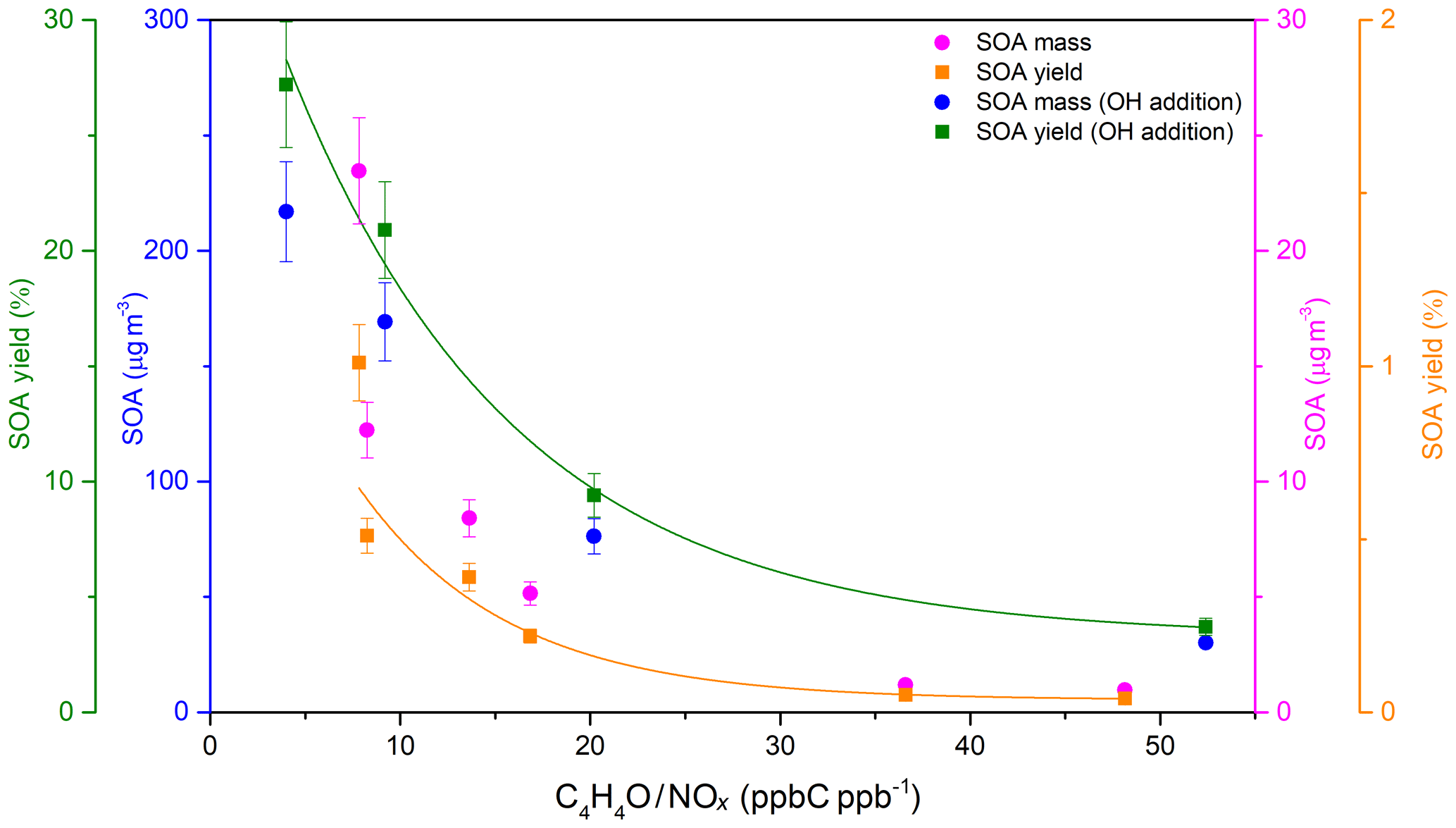
Figure 3Dependence of the SOA mass concentration and SOA yield on the C4H4O∕NOx ratio. Since the particle wall loss has a weak RH dependence in our chamber, a mean value of s−1 was used for wall loss correction. A density of 1.4 g cm−3 was used in SMPS (Jia and Xu, 2018; Kostenidou et al., 2007).

Figure 4Dependences of the SOA mass concentration, SOA yield, and ALW on relative humidity (RH).
It is generally accepted that experiments with low NOx levels lead to higher SOA yields than those with higher NOx levels at the same VOC concentration (Song et al., 2005). However, as shown in Fig. 3, an increasing SOA mass concentration and SOA yield with increasing NOx was observed. There are two possible explanations for this phenomenon: (i) the concentration of OH radicals produced in situ in the present study before an additional source of OH was insufficient to produce a considerable amount of SOA under low-NOx conditions. As shown in Fig. S3, the OH concentration exhibits a gradual increase with NOx concentration and there appears to be a correlation between NOx concentration, OH concentration and SOA yield. Therefore, at low-NOx conditions, the increase in SOA yield was attributed to an increase in OH concentration, which was affected by OH recycling following Reaction (R6) (see Fig. 1) and contributed to the enhancement of SOA formation. This result is consistent with a previous study concerning the impact of NOx and OH on SOA formation from β-pinene photooxidation, which has proved that the positive correlation between SOA yield and NOx levels ( ppbC ppb−1) was caused by the NOx-induced increase in OH concentration (Sarrafzadeh et al., 2016). (ii) Differently, Sarrafzadeh et al. found that after eliminating the effect of OH concentration on SOA mass growth, SOA yield only decreased with increasing NOx levels (Sarrafzadeh et al., 2016). To further investigate the NOx effect on furan-generated SOA formation under adequate OH conditions, four more experiments (see Table S1) were carried out with an additional injection of H2O2 as the OH radical source before the start of each experiment. The SOA yield trend at different C4H4O∕NOx ratios is also shown in Fig. 3. The continuous growth trend of SOA yield with increasing NOx concentration at a relatively high NOx level may result from the partitioning of generated semi- or low-volatility compounds (multifunctional nitrates and dinitrates) into the particle phase, leading to significant furan SOA formation under high-NOx conditions. Similarly, SOA from OH-initiated isoprene oxidation under high-NOx conditions was comprehensively investigated by Schwantes et al. (2019), who suggested that low-volatility hydroxyl nitrates and dihydroxyl dinitrates generated conspicuously more aerosol than previously thought (Schwantes et al., 2019). Our results showing the increase in SOA mass formation by high-NOx conditions also agree with a previous study, which indicated that a high level of NO2 can participate in the OH-induced reaction of guaiacol, consequently leading to the formation of organic nitrates and the enhancement of guaiacol SOA formation (C. Liu et al., 2019).
It is worth noting that under high-RH conditions, as shown in Fig. 1, the NO2 hydrolysis (Reaction R11) can generate nitrous acid (HONO), which has been regarded as a major source of OH. As indicated in Fig. 4, the SOA yields obtained in the present work clearly show a gradual increase with RH. Also shown in Fig. S3 is the dependence of OH and furan concentrations on RH during the experiments determined from the decay of furan using a reaction rate coefficient of k(OH + furan) = cm3 molecules−1 s−1 (Atkinson et al., 1983). It is therefore probable that the increase in RH results in high levels of HONO formation in the chamber, which leads to an increase in OH concentration, a faster furan decay rate, and higher aerosol mass yields. This result is in reasonably good agreement with previous studies, which proposed that the amount of products that can partition into the particle phase increases with the increasing rate of hydrocarbon oxidation (Healy et al., 2009; Chan et al., 2007). Moreover, the increasing RH might also enhance the SOA formation due to the fact that the functionalized gas-phase components were more favoured to condense on the surface of wet particles (S. Liu et al., 2019).
3.2 SOA chemical composition
To get detailed information on the functional groups in SOA formed during the photooxidation of furan, the collected particles were measured by FTIR, which has been proven to be an ideal technique for the detection of functional groups and bond information in aerosol samples. The FTIR spectra of particles collected from furan–NOx–NaCl with different values of C4H4O∕NOx ratio are shown in Fig. 5. The absorptions of organic functional groups were detected, which further confirmed the SOA formation from the photooxidation of furan. The assignment of the FTIR absorption frequencies is summarized in Table 2. The organic nitrate exhibits typical NO symmetric stretching at 868 cm−1, NO2 symmetric stretching at around 1341 cm−1, and NO2 asymmetric stretching at 1614 cm−1 (Jia and Xu, 2016). The absorption at 1067 cm−1 matches the C-O stretching vibration in C-O-C, while the sharp absorption at 1724 cm−1 is the C=O stretching vibration in carboxylic acid and ketones (Sakamoto et al., 2013). The carbon skeleton corresponds to the vibrations between 2850 and 3100 cm−1, where the ν(C-H) stretching vibration can be found. Absorptions in the ranges 2850–3000 and 3000–3100 cm−1 represent the C-H stretching vibration in saturated carbon ring and unsaturated alkenes, respectively. Correspondingly, the C-H bending vibrations are represented by the absorption between 1350 and 1515 cm−1. The strong broad vibrations at 2400–3500 cm−1 are interpreted as the O-H stretching vibration in carboxyl groups and hydroxyl groups (Ge et al., 2017a).
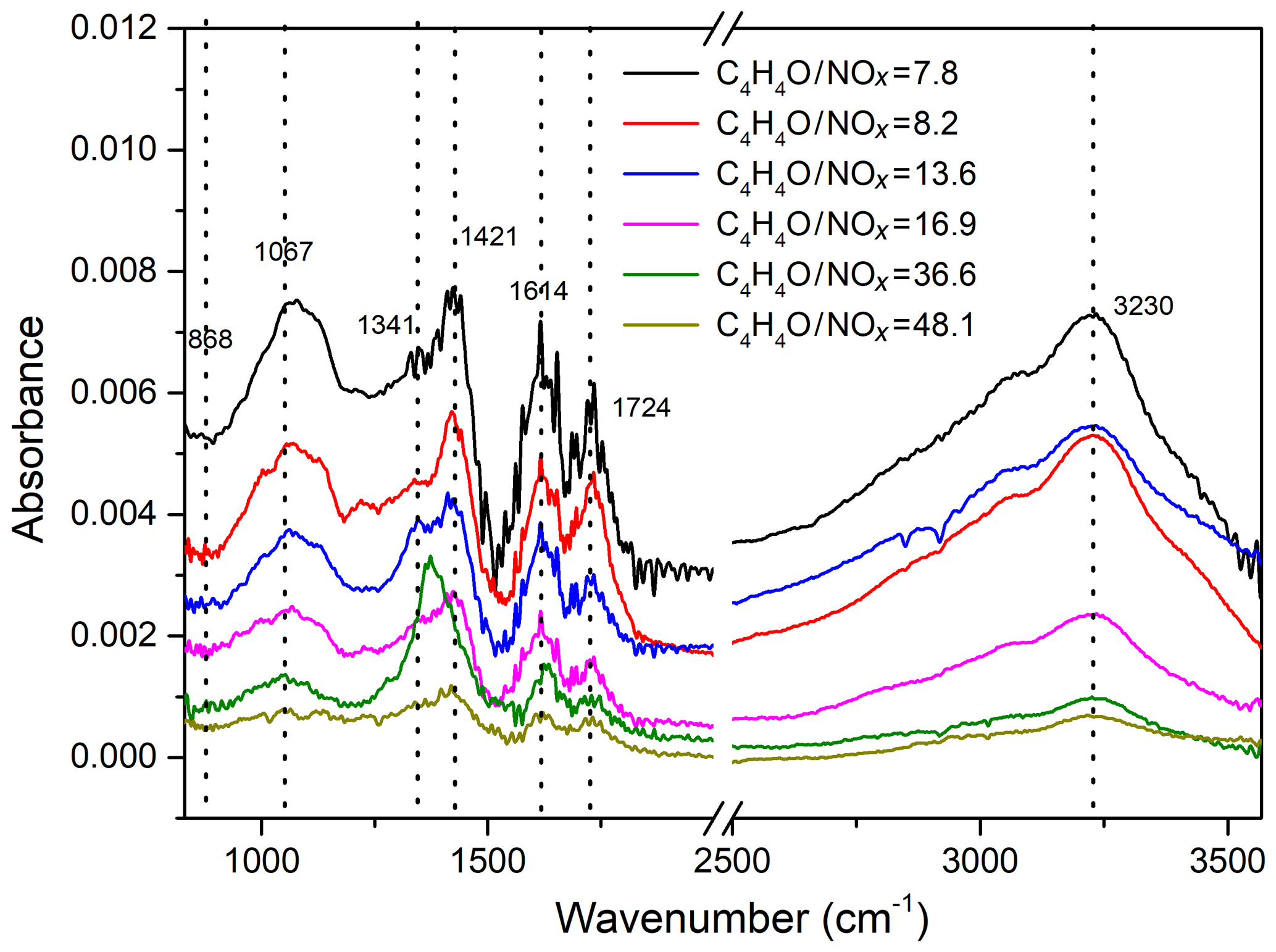
Figure 5FTIR spectra of particles collected from furan–NOx–NaCl photooxidation experiments with different values of C4H4O∕NOx ratio, ranging from 7.3 to 48.1.
Table 2Assignment of the observed FTIR absorption frequencies (cm−1).

Figure 5 illustrates the influence of NOx level on the SOA components during the photooxidation of furan. For each of the functional groups, experiments with the most pronounced SOA formation are those with the lowest initial C4H4O∕NOx ratio. This is further confirmed by comparing the SOA yield for different NOx level conditions under approximately similar gas-phase and initial seed conditions. The calculated variations of the relative abundance of FTIR functional groups at different NOx levels are presented in Fig. S4, in which the absolute abundances are normalized with respect to the corresponding functional abundance detected at C4H4O∕NOx = 7.8. The carbonyl compound formed with a 7.8 initial C4H4O∕NOx ratio is approximately three times more abundant than that formed with a 48.1 initial C4H4O∕NOx ratio. The absorbance of NO2 functional groups exhibits a much stronger enhancement under an initial C4H4O∕NOx ratio of 7.8 compared to 48.1. Although at different C4H4O∕NOx ratio conditions the intensities of C-O-C and O-H functional groups show similar trends, their variations are substantially different from the variations of other functional groups. In sum, the increased absorbance of functional groups with decreasing initial C4H4O∕NOx ratios demonstrates that a relatively high NOx level contributes to the formation of SOA.
The FTIR results in the investigation of furan photooxidation under different RH conditions are shown in Fig. 6. The absorbance of the FTIR characteristic peak increases with the RH rising from 5 % to 85 %. Note that obvious intensities of the functional groups were observed when the RH exceeded the efflorescence RH of NaCl. In contrast, the absorptions of corresponding functional groups enhance gently when the RH is lower than 42 %. This phenomenon is consistent with the results of the SOA yield discussed above and can also be interpreted as the increasing ALW components contributing to SOA formation. The ratio of the absorbance intensities detected at low RH to that at 85 % RH is used as the relative abundance to show more intuitive FTIR results. As shown in Fig. S5, for RH between 5 % and 37 %, the intensities of all functional groups vary weakly and are approximately one-third of the intensity at 85 % RH. However, when the RH rises from 42 % to 85 %, the absorption intensities of O-H and NO2 functional groups increase by factors of 2.0 and 1.9, respectively. In this RH range, the variations of relative intensities are even stronger for C=O, C-H, and C-O-C, being increased by factors of 2.3, 2.3, and 2.5, respectively. A previous study has observed that in an urban environment containing aromatic hydrocarbons and NOx, the SOA yield increased by a factor of 2 to 3 under high RH compared to lower RH (Zhou et al., 2011). This trend is similar to that found in experiments with toluene in an urban aromatic hydrocarbon–NOx mixture system (Kamens et al., 2011). It should be pointed out that observed FTIR results show a high degree of consistency with the SOA mass concentration and SOA yield.
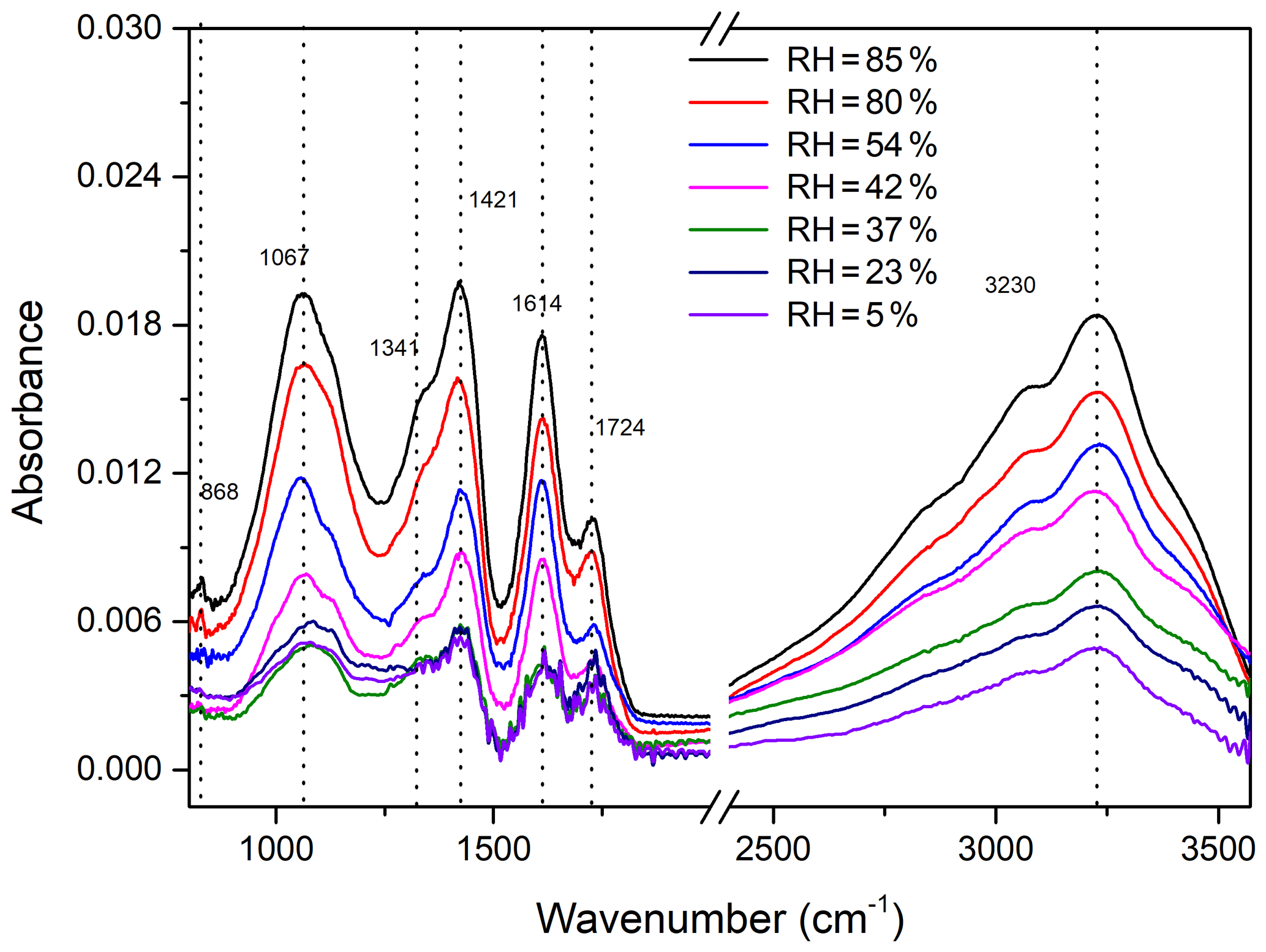
Figure 6FTIR spectra of particles collected from furan–NOx–NaCl photooxidation experiments with different values of RH, ranging from 5 % to 85 %.
To further identify and confirm the structure of generated SOA components, more techniques, such as GC/MS and HESI-Q Exactive-Orbitrap MS, were used to analyse the chemical composition. The chromatography before electrospray ionization (ESI)-MS analyses is helpful to remove the disturbances of the inorganic salts and determine the appropriate molecular compound (Surratt et al., 2007; Gao et al., 2006). However, the collected PM products were not sufficient for the chromatography owing to the limitation of the chamber volume. Furthermore, the low SOA yield of the furan photooxidation make it harder to retain enough SOA components for the better response of ESI-MS signal. Consequently, direct infusion analyses were carried out for HESI-Q Exactive-Orbitrap MS in the present study. Five experiments of furan–NOx–NaCl photooxidation that were conducted under different initial C4H4O∕NOx ratios and RH conditions were analysed by HESI-Q Exactive-Orbitrap MS. The mass spectra recorded in different ion modes represent the detected compounds ionizable in either positive or negative modes (Walser et al., 2008). The MS spectra of generated species from different NOx level and RH conditions, which show evidence for the OH-furan reaction, are presented in Figs. 7 and 8, respectively. The major peaks are 85.0018, 101.0894, and 185.0504 in the positive ion mode and 146.0161, 225.0125, and 263.0132 in the negative ion mode. The prominent peaks in the HR-MS spectra detected in negative ion mode are comprised of various functionalized hydroxyl nitrates and dihydroxyl dinitrates. However, in the positive ion mode analysis, most carbonyl components were detected. The assignments of these ion peaks, the molecular weights of the products observed, and proposed structures are summarized in Table 3. These detected compounds provide additional evidence for the proposed radical reaction mechanism.
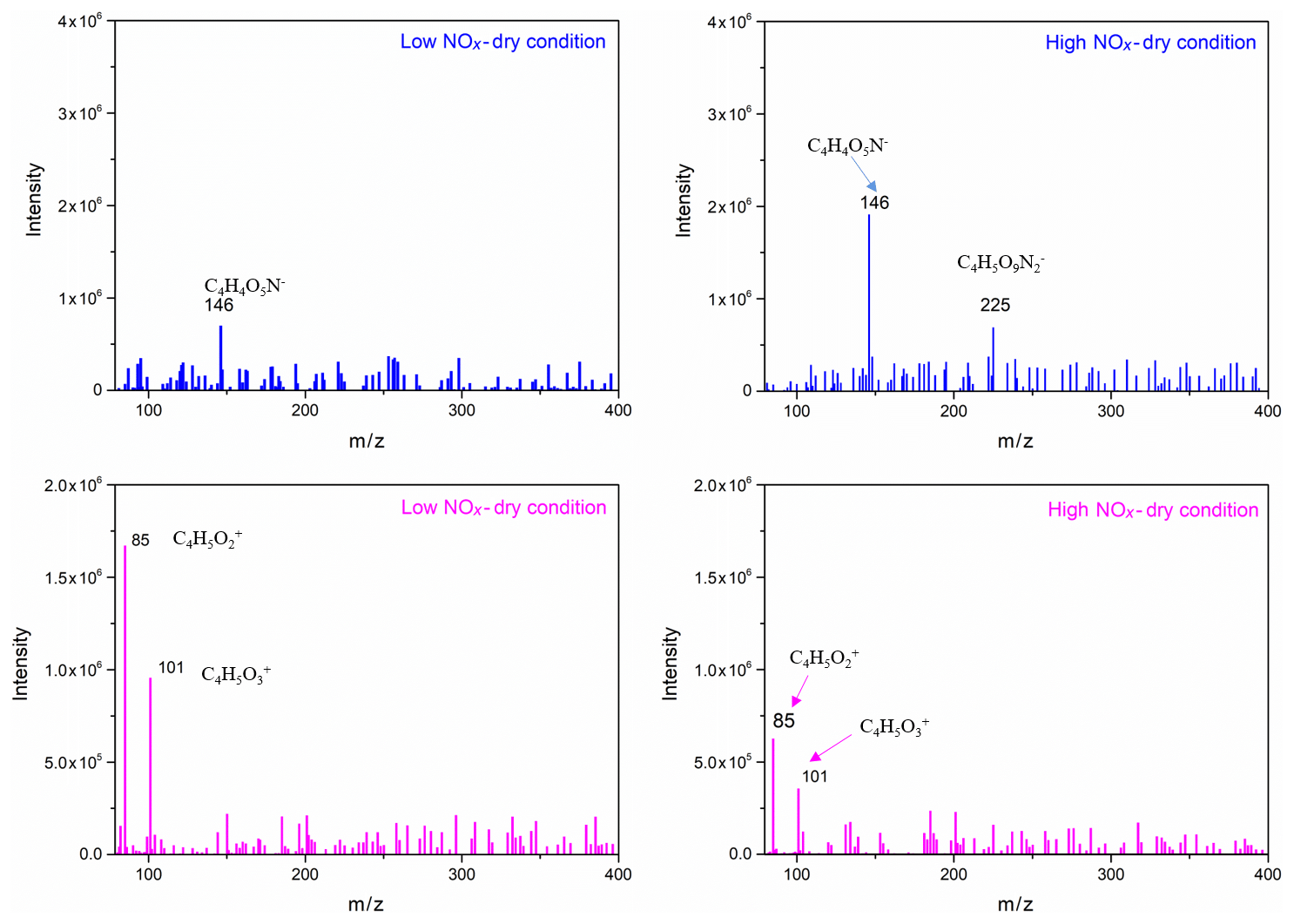
Figure 7Selected background-subtraction HESI-Q Exactive-Orbitrap MS results of SOA in both negative (blue) and positive (pink) ion modes from the photooxidation of furan under different RH conditions.
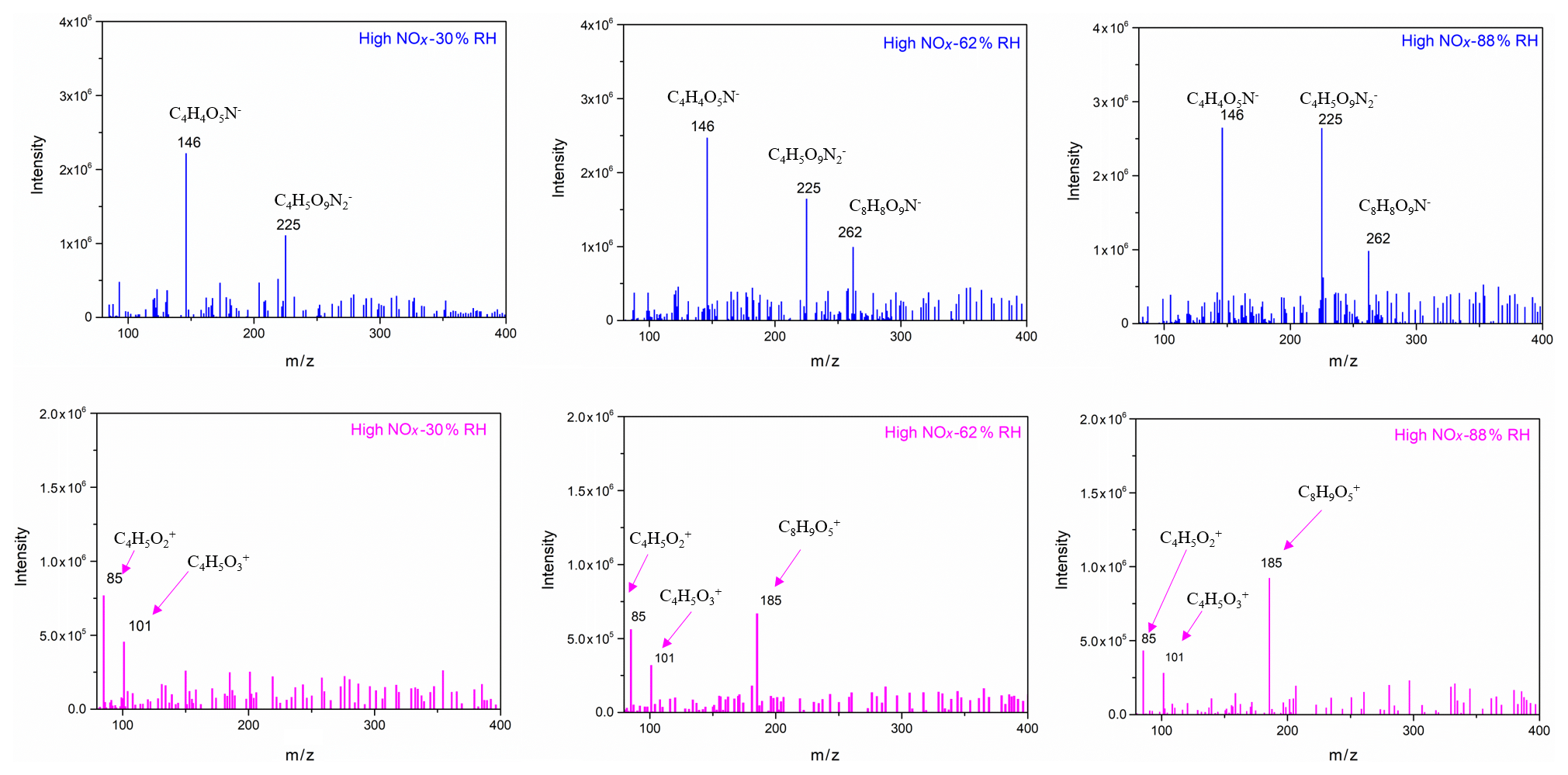
Figure 8Selected background-subtraction HESI-Q Exactive-Orbitrap MS results of SOA in both negative (blue) and positive (pink) ion modes from the photooxidation of furan under different RH conditions.
Table 3Ion peaks with the assigned compounds observed in the HESI-Q Exactive-Orbitrap MS. Proposed assignments are based on the formula from HESI-Q Exactive-Orbitrap MS.

According to the identified products in this work and based on previous kinetic (Atkinson et al., 1983; Lee and Tang, 1982) and product (Villanueva et al., 2009, 2007; Aschmann et al., 2014; Tapia et al., 2011; Strollo and Ziemann, 2013) studies, a proposed chemical mechanism for SOA formation from furan in the presence of NOx is shown in Scheme 1. Additionally, on the basis of well-established mechanisms for atmospheric volatile organic compounds (Atkinson and Arey, 2003), it can be concluded that the reaction is initiated by OH addition to a C=C bond at C2 or C3 positions. Addition at the C2 position forms two cyclic alkyl radicals (a, c), one of which (c) can isomerize to form a ring-opened alkyl radical (d), whereas addition at the C3 position forms a single alkyl radical (b). The OH radical addition leading to a hydrogen abstraction generates the alkyl radicals (R•) followed by reaction with O2 to form alkylperoxy radicals (RO2•) (Pan and Wang, 2014). Moreover, the formed alkylperoxy radicals (RO2•) can either react with RO2∕HO2 or NO to yield the corresponding alkoxy radical (RO•), which can (i) decompose and then react with O2 to yield a 1,4-aldoester (A), (ii) react with to form hydroxyl nitrate compound isomers with 146, or (iii) react with O2 to form unsaturated products and hydroxyfuranone (B) and 1,4-aldoacid (C). The ring-opened alkylperoxy radical generated from (d) can decompose to generate an unsaturated 1,4-dialdehyde (D). The formation of 1,4-dialdehyde with 85 detected in the positive ion mode suggests that these unsaturated 1,4-dicarbonyls are formed after initial OH addition at 2- or 5-positions. We note that OH radical addition at 2, 3-positions would lead to carbonyl product isomers with the same 101. In addition, some dihydroxyl dinitrates with 225 were also detected in negative ion mode. However, the pathways favouring the generation of these dihydroxyl dinitrates could only take place under high NOx levels. Scheme 1 shows the formation of second-generation products hemiacetals (E) via the reactions of the hydroxyfuranone (B) with 1,4-dialdehyde (D). After uptake from the gas phase, the combination of hydroxyfuranone with 4-dialdehyde appeared to occur by H abstraction, followed by dehydration, thus forming 185 compounds. This reaction pathway has also been identified in previous studies of OH-initiated reactions of furans (Strollo and Ziemann, 2013; Aschmann et al., 2014). According to the results of HR-MS, this aqueous-phase reaction is more favoured in aqueous particles.
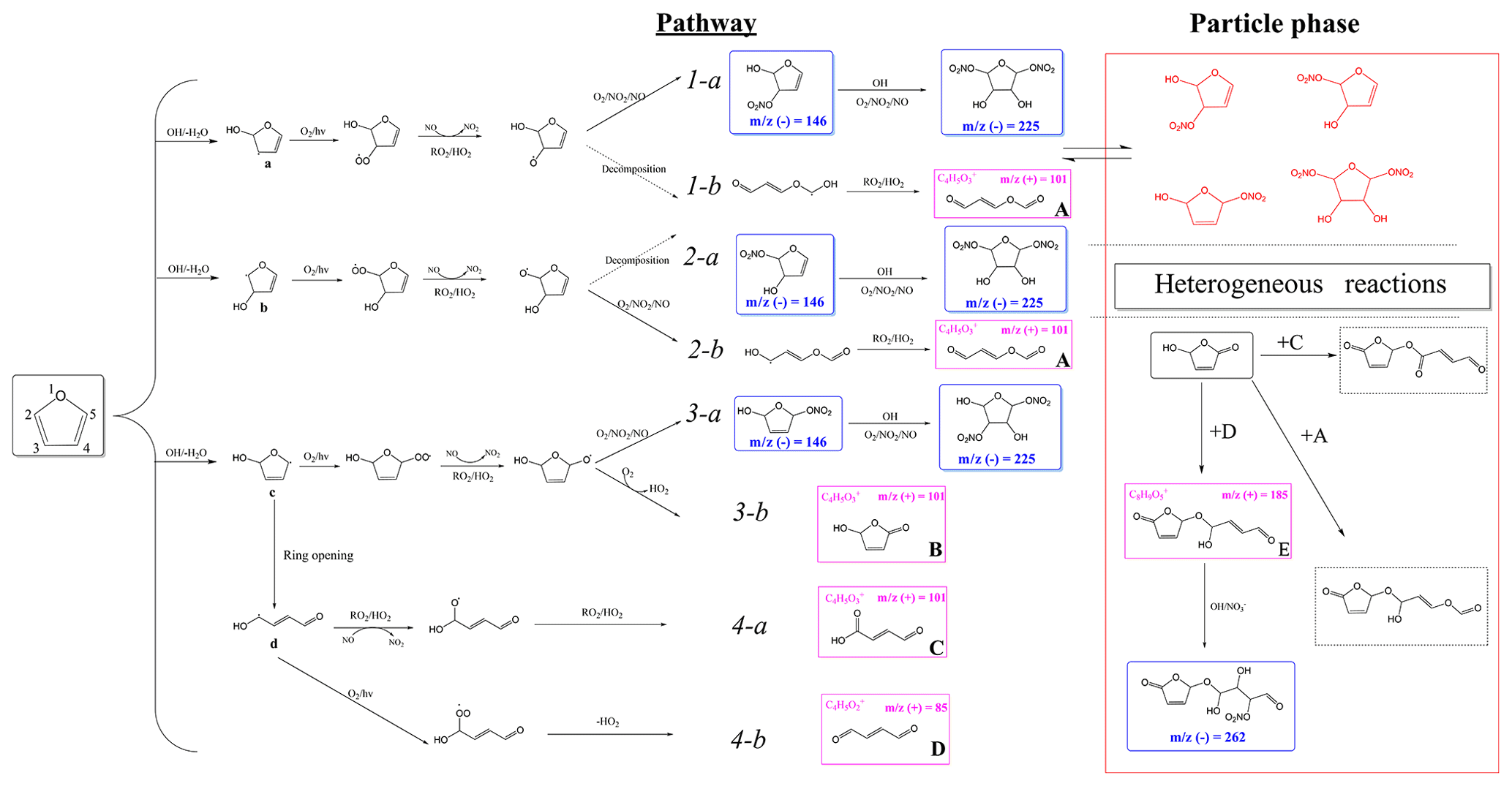
Scheme 1Proposed chemical mechanism of furan–NOx photooxidation under different experimental conditions. SOA constituents in blue and pink boxes are proposed SOA constituents detected by HESI-Q Exactive-Orbitrap MS under negative and positive ion mode, respectively. The detected nitrates and dinitrates in red are low-volatility organic species, which can easily partition into the particle phase and enhance the SOA formation.
3.3 Effects of NOx on SOA formation
To study the NOx level effect on SOA formation from the photooxidation of furan, the experiments were conducted with varying initial C4H4O∕NOx ratios ranging from 7.8 to 48.1. The SOA formation is found to have much lower yield under a high C4H4O∕NOx ratio. The SOA mass concentration and SOA yield increased from 1.0 to 23.5 µg m−3 and 0.04 % to 1.01 %, respectively, as the initial concentration ratio of C4H4O∕NOx decreased from 48.1 to 7.8 (ppbC ppb−1). This trend is consistent with previous studies on propylene photooxidation, which found that the SOA yield was enhanced under a low VOC∕NOx ratio (Ge et al., 2017a). Another laboratory study of aerosol assessment from isoprene photooxidation concluded that under a low NOx level (<129 ppb), the SOA mass increased with increasing initial NOx level (Kroll et al., 2006). By comparing the studies of Ge et al. (2017a) and Kroll et al. (2006), there is a positive correlation between NOx effect and SOA formation when the initial NOx level is relatively low (nearly below 100 ppb). These results further support our findings in the experiments of furan photooxidation. As shown in Fig. S6, SOA formed under a low C4H4O∕NOx ratio also have a faster increase than those under large C4H4O∕NOx ratio conditions. However, the particle number distribution under and conditions shows similar profiles.
Generally, the NOx level has two different effects in the reaction process. Firstly, increasing NOx concentration will promote the O3 and HONO formation, leading to more OH radical formed, which in turn is favourable to SOA formation (Sarrafzadeh et al., 2016). In addition, sufficient NOx can facilitate the competition between NO and HO2 to react with RO2. Products with high volatility will be generated more by the NO+RO2 reaction than by the HO2+RO2 reaction (Kroll and Seinfeld, 2008). However, the formation of lower-volatility products favours the increase in SOA yields (Chen et al., 2018). In this regard, the increasing NOx level is not conducive to SOA formation. It was shown that the yields of SOA generated from the photooxidation of m-xylene increased firstly and then decreased with the increase in the NOx level (Chen et al., 2018). In the present study, an increasing trend of SOA formation was observed with the increase in NOx concentration. As shown in the HESI-Q Exactive-Orbitrap MS results, all the detected primary products are carbonyl-rich, and even the organonitrates have at least two carbonyl functional groups. These carbonyl-containing products have lower volatility and contribute to the SOA formation. The peak intensities in the MS of the products ( 85, 101) generated by the pathways involving HO2 (as indicated by Scheme 1) decreased with the increase in NOx concentration. Additionally, more products of dihydroxyl dinitrates ( 225) with multifunctional groups were detected under high-NOx conditions. As shown in Scheme 1, the multifunctional organonitrates detected in negative ion mode are produced mostly from later-generation chemistry. Hydroxyl nitrates with 146 can be formed through pathways 1-a, 2-a, and 3-a by the reaction of RO2 with NOx. We note that the 146 compound was detected both at low NOx levels and high NOx levels. However, the peak intensity of this product was decreased with increasing NOx concentration. This phenomenon might be caused by the later reaction of the unsaturated hydroxyl nitrates going through a second OH-initiated reaction and leading to the formation of the dihydroxyl dinitrate with 225. In addition, the peak intensity changes of SOA products detected in the positive mode, such as the peaks at 85 and 101, were reduced under high-NOx conditions, which resulted from the fact that the RO2 radical fate was dominated by the pathway of RO2+NO or RO2+NO2. This result supports that the fate of RO2 is not a single channel reaction. There exists a competition between RO2 reacting with NOx and with HO2 under high-NOx conditions, but the former pathway is more favourable. There are two pathways for hydroxyl nitrates formation from RO2 radicals in the presence of NOx according to which RO2 radicals may react with NO and NO2 to form RONO2 and ROONO2, respectively (Kroll and Seinfeld, 2008). However, the formed peroxynitrates could easily thermally dissociate and convert to RONO2.
Furthermore, by analysing the OH concentration and product components, we conclude that there are two possible explanations for the increasing trend of SOA yield as the NOx level increases: (i) the SOA production is closely related to the oxidation capacity in the photooxidation experiments. Experiments conducted under different NOx levels indicate that the OH concentration is controlled by the NOx level if there is no additional OH precursor added before the start of the experiment. As shown in Fig. S3, an increase in NOx level results in more OH generation and a faster furan decay rate. This justifies the observed higher SOA mass concentration and higher SOA yield. (ii) HRMS fragments associated with multifunctional organonitrates are enhanced under high-NOx conditions (Fig. 7). As presented in Scheme 1, the furan dihydroxyl dinitrates are generated from the first-generation hydroxyl nitrate reacting with OH to form a peroxy radical, which reacts thereafter with NO. Together with multifunctional hydroxyl nitrates, these low-volatility species can easily partition into the particle phase and increase the SOA mass concentration. More importantly, the seed particle added initially plays an important role in the processes of gas-particle partitioning as indicated by a recent study, which showed that sufficient seed surface area at the start of the reaction largely suppressed the effects of vapour wall losses of low-volatility compounds (Schwantes et al., 2019). Therefore, the NaCl seed particle added in the present work promoted the partitioning of the formed low-volatility functional organonitrates.
3.4 Effect of RH on SOA formation
Experiments 6–12 were conducted under seven different RH conditions ranging from 5 % to 85 %. In this RH range, the SOA yield increases from 1.01 % to 5.03 %. With almost identical initial conditions except RH, the yield of furan-derived SOA formed at high RH can be a factor of 2 higher than that formed at low RH. A similar trend was also observed by Yu et al. (2011), who found that the SOA mass concentrations increased by a factor of 6 when RH increased from 18 % to 82 % (Yu et al., 2011). As shown in Fig. S3, an increase in RH leads to higher OH concentration resulting from higher HONO levels generated by the reaction of NO2with H2O. Previously, using quantum mechanical calculations, Anglada et al. (2011) confirmed that the water component could increase the OH production. The positive correlation between initial water vapour concentration and OH concentration has also been previously observed experimentally (Healy et al., 2009; Tillmann et al., 2010). Additionally, Healy et al. (2009) have also reported that increasing OH concentration promoted the decay of VOC and enhanced SOA formation (Healy et al., 2009). Similarly, in the present work, a faster decay rate of furan was also observed as RH increased, as shown in Fig. S3. It is possible that the faster rate of gas-phase oxidation under higher OH concentrations will lead to the generation of less volatile compounds as presented previously (Chan et al., 2007). A higher OH concentration promotes oxidation reactions, influences the distribution of organic products, and facilitates the SOA formation (Sarrafzadeh et al., 2016).
An obvious increase in SOA yield was observed when the RH increased from 37 % to 54 %. This phenomenon was mainly caused by the efflorescence transition when the seed particles were coated with SOA. It has been previously shown that SOA formation decreases both the efflorescence RH and deliquescence RH of the seed particles and results in the uptake of water by the particles (Liu et al., 2018; Takahama et al., 2007; Smith et al., 2012). It is highly possible that the NaCl seeds effloresce and deliquesce early after being coated by the newly formed SOA. The effect of efflorescence contributes to the water uptake by the particles, leading to the obvious trend changing of SOA yield. It is noted that with the NaCl seed aerosols serving as nuclei, the ALW was high at high RH. Products with water solubility produced from the photooxidation of furan can dissolve into the ALW of aerosol particles. As a result, ALW in the formed aerosols plays an important role in gas or particle partitioning. As shown in Fig. 4, the ALW was detected when the RH was higher than 54 %, which was based on the deliquescence of NaCl under high-RH conditions. The increase in ALW could partially explain the increase in SOA mass concentration and SOA yield. It is highly probable that the particle surface area increases with an increasing amount of ALW as shown in Fig. S7, which likely promotes the dissolution of semi-volatile matters produced during the experiments. According to the HESI-Q Exactive-Orbitrap MS results shown in Fig. 8, the intensities of multifunctional hydroxyl nitrates and dihydroxyl dinitrate ( 146 and 225, respectively) exhibited positive correlations with RH. Slight peak intensity increases in 85 and 101 products were also observed under high-RH conditions. This phenomenon indicates that the gas-particle phase partitioning of low-volatility compounds was enhanced under high-RH conditions. Furthermore, the increased surface area under high-RH conditions may also be attributed to the condensation of the produced multifunctional compounds.
Another possibility for the increasing trend of SOA yield with the increase in RH might result from the SOA formation through aqueous chemistry in wet aerosols (Grgic et al., 2010; Lim et al., 2010). In these atmospheric processes, alcohols, aldehydes, and ketones formed from the photooxidation of furan in the gas phase can be absorbed into the humid surface of the hygroscopic SOA at high RH. This process further contributes to the formation of low-volatility products on the SOA surface. In addition, the aqueous photochemistry of highly soluble small compounds that partitioned in ALW could produce additional organic compounds and result in a larger SOA yield under high-RH conditions (Faust et al., 2017; Jia and Xu, 2014). The appearance of 185 and 262 detected by the HESI-Q Exactive-Orbitrap MS further demonstrated that aqueous-phase reactions indeed took place under high-RH conditions. As shown in Scheme 1, the peak of 185 could form by aqueous-phase reaction of the hydroxyfuranone (B) and 1,4-dialdehyde (D). The formation of hemiacetal (E) has also been detected by a previous study of the OH-initiated reaction of 3-methylfuran in the presence of NOx (Strollo and Ziemann, 2013). The proposed hemiacetal compound (E) plays a substantial role in the obvious increase in 185 product formation under high-RH conditions. A pathway of organonitrate ( 262) formation in the aqueous particles with the presence of was suggested in the present study based on a previous work, which indicated a radical–radical reaction pathway for organosulfate formation from aqueous OH oxidation of glycolaldehyde in the presence of sulfuric acid (Perri et al., 2010). Increasing the RH also resulted in an overall addition of peak intensities in the negative ion mode, due to the fact that the sample obtained at high RH during the SOA generation had a larger particle surface. Specifically, a relatively stronger intense band of C8H8O9N− compound () was found under a high RH. Consequently, the heterogeneous products in wet seed particles will further contribute to the formation of SOA because higher aerosol liquid water content enables more aqueous-phase reactions.
In conclusion, the reasons for the increasing trend of SOA formation under high-RH conditions may be summarized as following: firstly, higher concentration of OH radical will certainly promote the SOA formation as RH increases. A faster decay rate of furan will also contribute to the formation of products that can partition into the particle phase. In addition, it is possible that the aqueous surface of seed particles provides a new substrate for the photooxidation of furan. Previously, N2O5 and HNO3 have been proven to be the key products in the VOC–NOx irradiation experiments (Wang et al., 2016). The moist surface under high-RH conditions is more favourable for the condensation of products with low vapour pressure, leading to the increasing production of SOA formation. The high-RH environment favours the formation of the hemiacetal compounds. Moreover, the effect of RH on SOA formation in furan photooxidation can also be determined by the aqueous photochemistry under high-RH conditions as discussed above. The aqueous-phase reactions at the surface of particles promote the formation of hemiacetal-like products, which likely plays an important role in the process of SOA formation. Previously, the unsaturated first-generation reaction product of 3-methyl furan has also been suggested to undergo acid-catalysed condensed-phase reactions, with SOA yields of up to 15 % (Strollo and Ziemann, 2013). In addition, the reinforced effect of RH on SOA yield was also ascribed from the photooxidation of other aromatic compounds, such as benzene (Ng et al., 2007b), toluene (Hildebrandt et al., 2009; Kamens et al., 2011), and xylene (Zhou et al., 2011).
The effects of NOx and RH on SOA formation from the photooxidation of furan in the presence of NaCl seed particles have been investigated in this study. The results demonstrated that the formation of SOA was promoted when the initial VOC∕NOx ratio decreases. The increase in SOA yield at a high VOC∕NOx ratio was caused by the NOx-induced increase in OH concentration. The reason for the promotion of SOA mass at a low furan ∕ NOx ratio is that more low-volatility products were generated from the furan photooxidation, which contributes to the formation of SOA. Additionally, the increase in RH results in the increase in the mass concentration of the produced SOA. The mechanisms controlling SOA formation may include the gas-phase photooxidation of furan, physical water uptake as RH increases, and the gas-phase reaction of water with the first-generation products and the aqueous chemistry of low-volatility products reacting at the wet surface of NaCl seed particles. Seed aerosols are important for the growth of atmospheric particles and, therefore, affect aerosol–cloud–climate interactions. Organic nitrates, detected with FTIR and ESI-Exactive-Orbitrap MS, were found to be significant in the composition of newly formed particles. A significant amount of carbonyl-rich products were also detected in the SOA products from the photooxidation of furan. A recent study showed that the reactive nitrogen chemistry in aerosol water can be a source of atmospheric sulfate during haze events (Cheng et al., 2016). In addition, the ALW is closely linked with air quality (Malm et al., 1994) and aqueous SOA formation (Sareen et al., 2017). The influence of the ALW component on SOA yield was examined and it was found that increased ALW amounts lead to higher SOA mass concentration and yield, therefore highlighting the importance of the ALW in photooxidation reactions. ALW plays a crucial role in atmospheric physicochemical processes. The current results could also be used to interpret ambient gas-phase measurements and reaction mechanism inference.
Data are available by contacting the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-13591-2019-supplement.
LD and XJ conceived and led the studies. XJ, NTT, LJ, SL, and HZ carried out the experiments and analysed the data. XJ, LD, and SL interpreted the results. NTT, LJ, and YX discussed the results and commented on the paper. XJ prepared the paper with contributions from all co-authors.
The authors declare that they have no conflict of interest.
This article is part of the special issue “Simulation chambers as tools in atmospheric research (AMT/ACP/GMD inter-journal SI)”. It is not associated with a conference.
We thank Hartmut Herrmann for his very constructive comments at the revision stage.
This research has been supported by the National Natural Science Foundation of China (grant nos. 91644214 and 41375129), the Shandong Natural Science Foundation for Distinguished Young Scholars (grant no. JQ201705), the Shandong Key R&D Program (grant no. 2018GSF117040), and the Fundamental Research Funds of Shandong University (grant no. 2017JQ01).
This paper was edited by Jason Surratt and reviewed by three anonymous referees.
Anglada, J. M., Gonzalez, J., and Torrent-Sucarrat, M.: Effects of the substituents on the reactivity of carbonyl oxides. A theoretical study on the reaction of substituted carbonyl oxides with water, Phys. Chem. Chem. Phys., 13, 13034–13045, https://doi.org/10.1039/c1cp20872a, 2011.
Aschmann, S. M., Nishino, N., Arey, J., and Atkinson, R.: Products of the OH radical initiated reactions of furan, 2- and 3-methylfuran, and 2,3- and 2,5-dimethylfuran in the presence of NO, J. Phys. Chem. A, 118, 457–466, https://doi.org/10.1021/jp410345k, 2014.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Atkinson, R., Aschmann, S. M., and Carter, W. P.: Kinetics of the reactions of O3 and OH radicals with furan and thiophene at 298±2 K, Int. J. Chem. Kinet., 15, 51–61, https://doi.org/10.1002/kin.550150106, 1983.
Atkinson, R., Aschmann, S. M., Winer, A. M., and Carter, W. P.: Rate constants for the gas-phase reactions of nitrate radicals with furan, thiophene, and pyrrole at 295±1 K and atmospheric pressure, Environ. Sci. Technol., 19, 87–90, https://doi.org/10.1021/es00131a010, 1985.
Baker, J., Arey, J., and Atkinson, R.: Formation and reaction of hydroxycarbonyls from the reaction of OH radicals with 1,3-butadiene and isoprene, Environ. Sci. Technol., 39, 4091–4099, https://doi.org/10.1021/es047930t, 2005.
Bonn, B. and Moorgat, G. K.: New particle formation during a- and b-pinene oxidation by O3, OH and NO3, and the influence of water vapour: particle size distribution studies, Atmos. Chem. Phys., 2, 183–196, https://doi.org/10.5194/acp-2-183-2002, 2002.
Bowman, F. M. and Seinfeld, J. H.: Fundamental basis of incremental reactivities of organics in ozone formation in VOC∕NOx mixtures, Atmos. Environ., 28, 3359–3368, https://doi.org/10.1016/1352-2310(94)00165-h, 1994.
Burling, I. R., Yokelson, R. J., Griffith, D. W. T., Johnson, T. J., Veres, P., Roberts, J. M., Warneke, C., Urbanski, S. P., Reardon, J., Weise, D. R., Hao, W. M., and de Gouw, J.: Laboratory measurements of trace gas emissions from biomass burning of fuel types from the southeastern and southwestern United States, Atmos. Chem. Phys., 10, 11115–11130, https://doi.org/10.5194/acp-10-11115-2010, 2010.
Cabanas, B., Baeza, M. T., Salgado, S., Martin, P., Taccone, R., and Martinez, E.: Oxidation of heterocycles in the atmosphere: Kinetic study of their reactions with NO3 radical, J. Phys. Chem. A, 108, 10818–10823, https://doi.org/10.1021/jp046524t, 2004.
Cao, G. and Jang, M.: An SOA model for toluene oxidation in the presence of inorganic aerosols, Environ. Sci. Technol., 44, 727–733, https://doi.org/10.1021/es901682r, 2010.
Chan, A. W. H., Kroll, J. H., Ng, N. L., and Seinfeld, J. H.: Kinetic modeling of secondary organic aerosol formation: effects of particle- and gas-phase reactions of semivolatile products, Atmos. Chem. Phys., 7, 4135–4147, https://doi.org/10.5194/acp-7-4135-2007, 2007.
Chan, A. W. H., Chan, M. N., Surratt, J. D., Chhabra, P. S., Loza, C. L., Crounse, J. D., Yee, L. D., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Role of aldehyde chemistry and NOx concentrations in secondary organic aerosol formation, Atmos. Chem. Phys., 10, 7169–7188, https://doi.org/10.5194/acp-10-7169-2010, 2010.
Chen, Y., Tong, S., Wang, J., Peng, C., Ge, M., Xie, X., and Sun, J.: The effect of Titanium Dioxide on secondary organic aerosol formation, Environ. Sci. Technol., 52, 11612–11620, https://doi.org/10.1021/acs.est.8b02466, 2018.
Cheng, Y., Zheng, G., Wei, C., Mu, Q., Zheng, B., Wang, Z., Gao, M., Zhang, Q., He, K., Carmichael, G., Poschl, U., and Su, H.: Reactive nitrogen chemistry in aerosol water as a source of sulfate during haze events in China, Sci. Rep.-UK, 2, e1601530, https://doi.org/10.1126/sciadv.1601530, 2016.
Cocker, D. R., Mader, B. T., Kalberer, M., Flagan, R. C., and Seinfeld, J. H.: The effect of water on gas-particle partitioning of secondary organic aerosol: II. m-xylene and 1,3,5-trimethylbenzene photooxidation systems, Atmos. Environ., 35, 6073–6085, https://doi.org/10.1016/s1352-2310(01)00405-8, 2001.
Coggon, M. M., Veres, P. R., Yuan, B., Koss, A., Warneke, C., Gilman, J. B., Lerner, B. M., Peischl, J., Aikin, K. C., Stockwell, C. E., Hatch, L. E., Ryerson, T. B., Roberts, J. M., Yokelson, R. J., and de Gouw, J. A.: Emissions of nitrogen-containing organic compounds from the burning of herbaceous and arboraceous biomass: Fuel composition dependence and the variability of commonly used nitrile tracers, Geophys. Res. Lett., 43, 9903–9912, https://doi.org/10.1002/2016gl070562, 2016.
Coury, C. and Dillner, A. M.: A method to quantify organic functional groups and inorganic compounds in ambient aerosols using attenuated total reflectance FTIR spectroscopy and multivariate chemometric techniques, Atmos. Environ., 42, 5923–5932, https://doi.org/10.1016/j.atmosenv.2008.03.026, 2008.
Donahue, N. M., Robinson, A. L., and Pandis, S. N.: Atmospheric organic particulate matter: From smoke to secondary organic aerosol, Atmos. Environ., 43, 94–106, https://doi.org/10.1016/j.atmosenv.2008.09.055, 2009.
Duarte, R., Pio, C. A., and Duarte, A. C.: Spectroscopic study of the water-soluble organic matter isolated from atmospheric aerosols collected under different atmospheric conditions, Anal. Chim. Acta, 530, 7–14, https://doi.org/10.1016/j.aca.2004.08.049, 2005.
Edney, E. O., Driscoll, D. J., Speer, R. E., Weathers, W. S., Kleindienst, T. E., Li, W., and Smith, D. F.: Impact of aerosol liquid water on secondary organic aerosol yields of irradiated toluene ∕ propylene ∕ NOx ∕ (NH4)2SO4 ∕ air mixtures, Atmos. Environ., 34, 3907–3919, https://doi.org/10.1016/s1352-2310(00)00174-6, 2000.
Engelhart, G. J., Hildebrandt, L., Kostenidou, E., Mihalopoulos, N., Donahue, N. M., and Pandis, S. N.: Water content of aged aerosol, Atmos. Chem. Phys., 11, 911–920, https://doi.org/10.5194/acp-11-911-2011, 2011.
Ervens, B., Turpin, B. J., and Weber, R. J.: Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies, Atmos. Chem. Phys., 11, 11069-11102, https://doi.org/10.5194/acp-11-11069-2011, 2011.
Faust, J. A., Wong, J. P. S., Lee, A. K. Y., and Abbatt, J. P. D.: Role of aerosol liquid water in secondary organic aerosol formation from volatile organic compounds, Environ. Sci. Technol., 51, 1405–1413, https://doi.org/10.1021/acs.est.6b04700, 2017.
Fry, J. L., Kiendler-Scharr, A., Rollins, A. W., Wooldridge, P. J., Brown, S. S., Fuchs, H., Dubé, W., Mensah, A., dal Maso, M., Tillmann, R., Dorn, H.-P., Brauers, T., and Cohen, R. C.: Organic nitrate and secondary organic aerosol yield from NO3 oxidation of β-pinene evaluated using a gas-phase kinetics/aerosol partitioning model, Atmos. Chem. Phys., 9, 1431–1449, https://doi.org/10.5194/acp-9-1431-2009, 2009.
Gao, S., Surratt, J. D., Knipping, E. M., Edgerton, E. S., Shahgholi, M., and Seinfeld, J. H.: Characterization of polar organic components in fine aerosols in the southeastern United States: Identity, origin, and evolution, J. Geophys. Res., 111, D14314, https://doi.org/10.1029/2005jd006601, 2006.
Ge, S., Xu, Y., and Jia, L.: Secondary organic aerosol formation from propylene irradiations in a chamber study, Atmos. Environ., 157, 146–155, https://doi.org/10.1016/j.atmosenv.2017.03.019, 2017a.
Ge, S., Xu, Y., and Jia, L.: Secondary organic aerosol formation from ethylene ozonolysis in the presence of sodium chloride, J. Aerosol Sci., 106, 120–131, https://doi.org/10.1016/j.jaerosci.2017.01.009, 2017b.
Gilman, J. B., Lerner, B. M., Kuster, W. C., Goldan, P. D., Warneke, C., Veres, P. R., Roberts, J. M., de Gouw, J. A., Burling, I. R., and Yokelson, R. J.: Biomass burning emissions and potential air quality impacts of volatile organic compounds and other trace gases from fuels common in the US, Atmos. Chem. Phys., 15, 13915–13938, https://doi.org/10.5194/acp-15-13915-2015, 2015.
Gloess, A. N., Vietri, A., Wieland, F., Smrke, S., Schoenbaechler, B., Lopez, J. A. S., Petrozzi, S., Bongers, S., Koziorowski, T., and Yeretzian, C.: Evidence of different flavour formation dynamics by roasting coffee from different origins: On-line analysis with PTR-ToF-MS, Int. J. Mass Spectrom., 365, 324–337, https://doi.org/10.1016/j.ijms.2014.02.010, 2014.
Gomez Alvarez, E., Borras, E., Viidanoja, J., and Hjorth, J.: Unsaturated dicarbonyl products from the OH-initiated photo-oxidation of furan, 2-methylfuran and 3-methylfuran, Atmos. Environ., 43, 1603–1612, https://doi.org/10.1016/j.atmosenv.2008.12.019, 2009.
Greenberg, J. P., Zimmerman, P. R., Heidt, L., and Pollock, W.: Hydrocarbon and carbon-monoxide emissions from biomass burning in Brazil, J. Geophys. Res., 89, 1350–1354, https://doi.org/10.1029/JD089iD01p01350, 1984.
Grgic, I., Nieto-Gligorovski, L. I., Net, S., Temime-Roussel, B., Gligorovski, S., and Wortham, H.: Light induced multiphase chemistry of gas-phase ozone on aqueous pyruvic and oxalic acids, Phys. Chem. Chem. Phys., 12, 698–707, https://doi.org/10.1039/b914377g, 2010.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Hatch, L. E., Luo, W., Pankow, J. F., Yokelson, R. J., Stockwell, C. E., and Barsanti, K. C.: Identification and quantification of gaseous organic compounds emitted from biomass burning using two-dimensional gas chromatography–time-of-flight mass spectrometry, Atmos. Chem. Phys., 15, 1865–1899, https://doi.org/10.5194/acp-15-1865-2015, 2015.
Healy, R. M., Temime, B., Kuprovskyte, K., and Wenger, J. C.: Effect of relative humidity on gas/particle partitioning and aerosol mass yield in the photooxidation of p-Xylene, Environ. Sci. Technol., 43, 1884–1889, https://doi.org/10.1021/es802404z, 2009.
Hildebrandt, L., Donahue, N. M., and Pandis, S. N.: High formation of secondary organic aerosol from the photo-oxidation of toluene, Atmos. Chem. Phys., 9, 2973–2986, https://doi.org/10.5194/acp-9-2973-2009, 2009.
Hinks, M. L., Montoya-Aguilera, J., Ellison, L., Lin, P., Laskin, A., Laskin, J., Shiraiwa, M., Dabdub, D., and Nizkorodov, S. A.: Effect of relative humidity on the composition of secondary organic aerosol from the oxidation of toluene, Atmos. Chem. Phys., 18, 1643–1652, https://doi.org/10.5194/acp-18-1643-2018, 2018.
IARC: Dry cleaning, some chlorinated solvents and other industrial chemical, IARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Humans, 63. International Agency for Research on Cancer, Lyon, France, 558 pp., 1995.
Jang, M. S., Czoschke, N. M., Lee, S., and Kamens, R. M.: Heterogeneous atmospheric aerosol production by acid-catalyzed particle-phase reactions, Science, 298, 814–817, https://doi.org/10.1126/science.1075798, 2002.
Jia, L. and Xu, Y.: Effects of relative humidity on ozone and secondary organic aerosol formation from the photooxidation of benzene and ethylbenzene, Aerosol Sci. Tech., 48, 1–12, https://doi.org/10.1080/02786826.2013.847269, 2014.
Jia, L. and Xu, Y.: Ozone and secondary organic aerosol formation from Ethylene-NO(x)-NaCl irradiations under different relative humidity conditions, J. Atmos. Chem., 73, 81–100, https://doi.org/10.1007/s10874-015-9317-1, 2016.
Jia, L. and Xu, Y.: Different roles of water in secondary organic aerosol formation from toluene and isoprene, Atmos. Chem. Phys., 18, 8137–8154, https://doi.org/10.5194/acp-18-8137-2018, 2018.
Jonsson, A. M., Hallquist, M., and Ljungstrom, E.: Impact of humidity on the ozone initiated oxidation of limonene, delta(3)-carene, and alpha-pinene, Environ. Sci. Technol., 40, 188–194, https://doi.org/10.1021/es051163w, 2006.
Kahan, T. F., Ormond, T. K., Ellison, G. B., and Vaida, V.: Acetic acid formation via the hydration of gas-phase ketene under ambient conditions, Chem. Phys. Lett., 565, 1–4, https://doi.org/10.1016/j.cplett.2013.02.030, 2013.
Kamens, R. M., Zhang, H., Chen, E. H., Zhou, Y., Parikh, H. M., Wilson, R. L., Galloway, K. E., and Rosen, E. P.: Secondary organic aerosol formation from toluene in an atmospheric hydrocarbon mixture: Water and particle seed effects, Atmos. Environ., 45, 2324–2334, https://doi.org/10.1016/j.atmosenv.2010.11.007, 2011.
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005.
Koch, B. P., Witt, M. R., Engbrodt, R., Dittmar, T., and Kattner, G.: Molecular formulae of marine and terrigenous dissolved organic matter detected by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry, Geochim. Cosmochim. Ac., 69, 3299–3308, https://doi.org/10.1016/j.gca.2005.02.027, 2005.
Kostenidou, E., Pathak, R. K., and Pandis, S. N.: An algorithm for the calculation of secondary organic aerosol density combining AMS and SMPS data, Aerosol Sci. Tech., 41, 1002–1010, https://doi.org/10.1080/02786820701666270, 2007.
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, https://doi.org/10.1016/j.atmosenv.2008.01.003, 2008.
Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from isoprene photooxidation, Environ. Sci. Technol., 40, 1869–1877, https://doi.org/10.1021/es0524301, 2006.
Labouze, E., Honoré, C., Moulay, L., Couffignal, B., and Beekmann, M.: Photochemical ozone creation potentials, Int. J. Life Cycle Ass., 9, 187–195, https://doi.org/10.1016/j.jclepro.2006.03.002, 2004.
Lee, J. H. and Tang, I. N.: Absolute rate constants for the hydroxyl radical reactions with ehtane, furan, and thiophene at roon temperature, J. Chem. Phys., 77, 4459–4463, https://doi.org/10.1063/1.444367, 1982.
Liljegren, J. A. and Stevens, P. S.: Kinetics of the reaction of OH radicals with 3-methylfuran at low pressure, Int. J. Chem. Kinet., 45, 787–794, https://doi.org/10.1002/kin.20814, 2013.
Lim, Y. B., Tan, Y., Perri, M. J., Seitzinger, S. P., and Turpin, B. J.: Aqueous chemistry and its role in secondary organic aerosol (SOA) formation, Atmos. Chem. Phys., 10, 10521–10539, https://doi.org/10.5194/acp-10-10521-2010, 2010.
Liu, C., Liu, J., Liu, Y., Chen, T., and He, H.: Secondary organic aerosol formation from the OH-initiated oxidation of guaiacol under different experimental conditions, Atmos. Environ., 207, 30–37, https://doi.org/10.1016/j.atmosenv.2019.03.021, 2019.
Liu, S., Jia, L., Xu, Y., Tsona, N. T., Ge, S., and Du, L.: Photooxidation of cyclohexene in the presence of SO2: SOA yield and chemical composition, Atmos. Chem. Phys., 17, 13329–13343, https://doi.org/10.5194/acp-17-13329-2017, 2017.
Liu, S., Tsona, N. T., Zhang, Q., Jia, L., Xu, Y., and Du, L.: Influence of relative humidity on cyclohexene SOA formation from OH photooxidation, Chemosphere, 231, 478–486, https://doi.org/10.1016/j.chemosphere.2019.05.131, 2019.
Liu, T., Huang, D. D., Li, Z., Liu, Q., Chan, M., and Chan, C. K.: Comparison of secondary organic aerosol formation from toluene on initially wet and dry ammonium sulfate particles at moderate relative humidity, Atmos. Chem. Phys., 18, 5677–5689, https://doi.org/10.5194/acp-18-5677-2018, 2018.
Mahowald, N., Ward, D. S., Kloster, S., Flanner, M. G., Heald, C. L., Heavens, N. G., Hess, P. G., Lamarque, J.-F., and Chuang, P. Y.: Aerosol impacts on climate and biogeochemistry, Annu. Rev. Env. Resour., 36, 45–74, 2011.
Malm, W. C., Sisler, J. F., Huffman, D., Eldred, R. A., and Cahill, T. A.: Spatial and seasonal trends in particle concentration and optical extinction in the United States, J. Geophys. Res., 99, 1347–1370, https://doi.org/10.1029/93jd02916, 1994.
Ng, N. L., Chhabra, P. S., Chan, A. W. H., Surratt, J. D., Kroll, J. H., Kwan, A. J., McCabe, D. C., Wennberg, P. O., Sorooshian, A., Murphy, S. M., Dalleska, N. F., Flagan, R. C., and Seinfeld, J. H.: Effect of NOx level on secondary organic aerosol (SOA) formation from the photooxidation of terpenes, Atmos. Chem. Phys., 7, 5159–5174, https://doi.org/10.5194/acp-7-5159-2007, 2007a.
Ng, N. L., Kroll, J. H., Chan, A. W. H., Chhabra, P. S., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from m-xylene, toluene, and benzene, Atmos. Chem. Phys., 7, 3909–3922, https://doi.org/10.5194/acp-7-3909-2007, 2007b.
Olsson, M.: Wheat straw and peat for fuel pellets – organic compounds from combustion, Biomass Bioenerg., 30, 555–564, https://doi.org/10.1016/j.biombioe.2006.01.005, 2006.
Pan, S. and Wang, L.: Atmospheric Oxidation Mechanism of m-Xylene Initiated by OH Radical, J. Phys. Chem. A, 118, 10778–10787, https://doi.org/10.1021/jp506815v, 2014.
Perri, M. J., Lim, Y. B., Seitzinger, S. P., and Turpin, B. J.: Organosulfates from glycolaldehyde in aqueous aerosols and clouds: Laboratory studies, Atmos. Environ., 44, 2658–2664, https://doi.org/10.1016/j.atmosenv.2010.03.031, 2010.
Perzon, M.: Emissions of organic compounds from the combustion of oats – a comparison with softwood pellets, Biomass Bioenerg., 34, 828–837, https://doi.org/10.1016/j.biombioe.2010.01.027, 2010.
Pope III, C. A., Ezzati, M., and Dockery, D. W.: Fine particulate air pollution and life expectancies in the United States: The role of influential observations, J. Air Waste Manage., 63, 129–132, https://doi.org/10.1080/10962247.2013.760353, 2013.
Sakamoto, Y., Inomata, S., and Hirokawa, J.: Oligomerization reaction of the criegee intermediate leads to secondary organic aerosol formation in ethylene ozonolysis, J. Phys. Chem. A, 117, 12912–12921, https://doi.org/10.1021/jp408672m, 2013.
Sareen, N., Waxman, E. M., Turpin, B. J., Volkamer, R., and Carlton, A. G.: Potential of aerosol liquid water to facilitate organic aerosol formation: Assessing knowledge gaps about precursors and partitioning, Environ. Sci. Technol., 51, 3327–3335, https://doi.org/10.1021/acs.est.6b04540, 2017.
Sarkar, C., Sinha, V., Kumar, V., Rupakheti, M., Panday, A., Mahata, K. S., Rupakheti, D., Kathayat, B., and Lawrence, M. G.: Overview of VOC emissions and chemistry from PTR-TOF-MS measurements during the SusKat-ABC campaign: high acetaldehyde, isoprene and isocyanic acid in wintertime air of the Kathmandu Valley, Atmos. Chem. Phys., 16, 3979–4003, https://doi.org/10.5194/acp-16-3979-2016, 2016.
Sarrafzadeh, M., Wildt, J., Pullinen, I., Springer, M., Kleist, E., Tillmann, R., Schmitt, S. H., Wu, C., Mentel, T. F., Zhao, D., Hastie, D. R., and Kiendler-Scharr, A.: Impact of NOx and OH on secondary organic aerosol formation from β-pinene photooxidation, Atmos. Chem. Phys., 16, 11237–11248, https://doi.org/10.5194/acp-16-11237-2016, 2016.
Schwantes, R. H., Charan, S. M., Bates, K. H., Huang, Y., Nguyen, T. B., Mai, H., Kong, W., Flagan, R. C., and Seinfeld, J. H.: Low-volatility compounds contribute significantly to isoprene secondary organic aerosol (SOA) under high-NOx conditions, Atmos. Chem. Phys., 19, 7255–7278, https://doi.org/10.5194/acp-19-7255-2019, 2019.
Shafizadeh, F.: Introduction to pyrolysis of biomass, J. Anal. Appl. Pyrol., 3, 283–305, https://doi.org/10.1016/0165-2370(82)80017-x, 1982.
Smith, M. L., Bertram, A. K., and Martin, S. T.: Deliquescence, efflorescence, and phase miscibility of mixed particles of ammonium sulfate and isoprene-derived secondary organic material, Atmos. Chem. Phys., 12, 9613–9628, https://doi.org/10.5194/acp-12-9613-2012, 2012.
Song, C., Na, K. S., and Cocker, D. R.: Impact of the hydrocarbon to NOx ratio on secondary organic aerosol formation, Environ. Sci. Technol., 39, 3143–3149, 10.1021/es0493244, 2005.
Song, C., Zaveri, R. A., Alexander, M. L., Thornton, J. A., Madronich, S., Ortega, J. V., Zelenyuk, A., Yu, X.-Y., Laskin, A., and Maughan, D. A.: Effect of hydrophobic primary organic aerosols on secondary organic aerosol formation from ozonolysis of alpha-pinene, Geophys. Res. Lett., 34, https://doi.org/10.1029/2007gl030720, 2007.
Spittler, M., Barnes, I., Bejan, I., Brockmann, K. J., Benter, T., and Wirtz, K.: Reactions of NO3 radicals with limonene and alpha-pinene: Product and SOA formation, Atmos. Environ., 40, S116–S127, https://doi.org/10.1016/j.atmosenv.2005.09.093, 2006.
Sprengnether, M., Demerjian, K. L., Donahue, N. M., and Anderson, J. G.: Product analysis of the OH oxidation of isoprene and 1,3-butadiene in the presence of NO, J. Geophys. Res., 107, 4268, https://doi.org/10.1029/2001jd000716, 2002.
Stockwell, C. E., Yokelson, R. J., Kreidenweis, S. M., Robinson, A. L., DeMott, P. J., Sullivan, R. C., Reardon, J., Ryan, K. C., Griffith, D. W. T., and Stevens, L.: Trace gas emissions from combustion of peat, crop residue, domestic biofuels, grasses, and other fuels: configuration and Fourier transform infrared (FTIR) component of the fourth Fire Lab at Missoula Experiment (FLAME-4), Atmos. Chem. Phys., 14, 9727–9754, https://doi.org/10.5194/acp-14-9727-2014, 2014.
Stockwell, C. E., Veres, P. R., Williams, J., and Yokelson, R. J.: Characterization of biomass burning emissions from cooking fires, peat, crop residue, and other fuels with high-resolution proton-transfer-reaction time-of-flight mass spectrometry, Atmos. Chem. Phys., 15, 845–865, https://doi.org/10.5194/acp-15-845-2015, 2015.
Stockwell, C. E., Jayarathne, T., Cochrane, M. A., Ryan, K. C., Putra, E. I., Saharjo, B. H., Nurhayati, A. D., Albar, I., Blake, D. R., Simpson, I. J., Stone, E. A., and Yokelson, R. J.: Field measurements of trace gases and aerosols emitted by peat fires in Central Kalimantan, Indonesia, during the 2015 El Niño, Atmos. Chem. Phys., 16, 11711–11732, https://doi.org/10.5194/acp-16-11711-2016, 2016.
Strollo, C. M. and Ziemann, P. J.: Products and mechanism of secondary organic aerosol formation from the reaction of 3-methylfuran with OH radicals in the presence of NOx, Atmos. Environ., 77, 534–543, https://doi.org/10.1016/j.atmosenv.2013.05.033, 2013.
Surratt, J. D., Kroll, J. H., Kleindienst, T. E., Edney, E. O., Claeys, M., Sorooshian, A., Ng, N. L., Offenberg, J. H., Lewandowski, M., Jaoui, M., Flagan, R. C., and Seinfeld, J. H.: Evidence for organosulfates in secondary organic aerosol, Environ. Sci. Technol., 41, 517–527, https://doi.org/10.1021/es062081q, 2007.
Takahama, S., Pathak, R. K., and Pandis, S. N.: Efflorescence transitions of ammonium sulfate particles coated with secondary organic aerosol, Environ. Sci. Technol., 41, 2289–2295, https://doi.org/10.1021/es0619915, 2007.
Tapia, A., Villanueva, F., Salgado, M. S., Cabañas, B., Martínez, E., and Martín, P.: Atmospheric degradation of 3-methylfuran: kinetic and products study, Atmos. Chem. Phys., 11, 3227–3241, https://doi.org/10.5194/acp-11-3227-2011, 2011.
Tillmann, R., Hallquist, M., Jonsson, Å. M., Kiendler-Scharr, A., Saathoff, H., Iinuma, Y., and Mentel, Th. F.: Influence of relative humidity and temperature on the production of pinonaldehyde and OH radicals from the ozonolysis of α-pinene, Atmos. Chem. Phys., 10, 7057–7072, https://doi.org/10.5194/acp-10-7057-2010, 2010.
Tuazon, E. C., Alvarado, A., Aschmann, S. M., Atkinson, R., and Arey, J.: Products of the gas-phase reactions of 1,3-butadiene with OH and NO3 radicals, Environ. Sci. Technol., 33, 3586–3595, https://doi.org/10.1021/es990193u, 1999.
Varutbangkul, V., Brechtel, F. J., Bahreini, R., Ng, N. L., Keywood, M. D., Kroll, J. H., Flagan, R. C., Seinfeld, J. H., Lee, A., and Goldstein, A. H.: Hygroscopicity of secondary organic aerosols formed by oxidation of cycloalkenes, monoterpenes, sesquiterpenes, and related compounds, Atmos. Chem. Phys., 6, 2367–2388, https://doi.org/10.5194/acp-6-2367-2006, 2006.
Villanueva, F., Barnes, I., Monedero, E., Salgado, S., Gómez, M. V., and Martin, P.: Primary product distribution from the Cl-atom initiated atmospheric degradation of furan: Environmental implications, Atmos. Environ., 41, 8796–8810, https://doi.org/10.1016/j.atmosenv.2007.07.053, 2007.
Villanueva, F., Cabañas, B., Monedero, E., Salgado, S., Bejan, I., and Martin, P.: Atmospheric degradation of alkylfurans with chlorine atoms: Product and mechanistic study, Atmos. Environ., 43, 2804–2813, https://doi.org/10.1016/j.atmosenv.2009.02.030, 2009.
Walser, M. L., Desyaterik, Y., Laskin, J., Laskin, A., and Nizkorodov, S. A.: High-resolution mass spectrometric analysis of secondary organic aerosol produced by ozonation of limonene, Phys. Chem. Chem. Phys., 10, 1009–1022, https://doi.org/10.1039/b712620d, 2008.
Wang, Y., Luo, H., Jia, L., and Ge, S.: Effect of particle water on ozone and secondary organic aerosol formation from benzene-NO2-NaCl irradiations, Atmos. Environ., 140, 386–394, https://doi.org/10.1016/j.atmosenv.2016.06.022, 2016.
Xu, J., Griffin, R. J., Liu, Y., Nakao, S., and Cocker III, D. R.: Simulated impact of NOx on SOA formation from oxidation of toluene and m-xylene, Atmos. Environ., 101, 217–225, https://doi.org/10.1016/j.atmosenv.2014.11.008, 2015.
Yu, K.-P., Lin, C.-C., Yang, S.-C., and Zhao, P.: Enhancement effect of relative humidity on the formation and regional respiratory deposition of secondary organic aerosol, J. Hazard. Mater., 191, 94–102, https://doi.org/10.1016/j.jhazmat.2011.04.042, 2011.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Ulbrich, I. M., Ng, N. L., Worsnop, D. R., and Sun, Y.: Understanding atmospheric organic aerosols via factor analysis of aerosol mass spectrometry: A review, Anal. Bioanal. Chem., 401, 3045–3067, https://doi.org/10.1007/s00216-011-5355-y, 2011.
Zhou, Y., Zhang, H., Parikh, H. M., Chen, E. H., Rattanavaraha, W., Rosen, E. P., Wang, W., and Kamens, R. M.: Secondary organic aerosol formation from xylenes and mixtures of toluene and xylenes in an atmospheric urban hydrocarbon mixture: Water and particle seed effects (II), Atmos. Environ., 45, 3882–3890, https://doi.org/10.1016/j.atmosenv.2010.12.048, 2011.