the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 14 Aug 2019
| 14 Aug 2019
Perfluorocyclobutane (PFC-318, c-C4F8) in the global atmosphere
Cathy M. Trudinger
Luke M. Western
Matthew Rigby
Martin K. Vollmer
Sunyoung Park
Alistair J. Manning
Daniel Say
Anita Ganesan
L. Paul Steele
Diane J. Ivy
Tim Arnold
Shanlan Li
Andreas Stohl
Christina M. Harth
Peter K. Salameh
Archie McCulloch
Simon O'Doherty
Mi-Kyung Park
Chun Ok Jo
Dickon Young
Kieran M. Stanley
Paul B. Krummel
Blagoj Mitrevski
Ove Hermansen
Chris Lunder
Nikolaos Evangeliou
Bo Yao
Jooil Kim
Benjamin Hmiel
Christo Buizert
Vasilii V. Petrenko
Jgor Arduini
Michela Maione
David M. Etheridge
Eleni Michalopoulou
Mike Czerniak
Jeffrey P. Severinghaus
Stefan Reimann
Peter G. Simmonds
Paul J. Fraser
Ronald G. Prinn
Ray F. Weiss
We reconstruct atmospheric abundances of the potent greenhouse gas c-C4F8 (perfluorocyclobutane, perfluorocarbon PFC-318) from measurements of in situ, archived, firn, and aircraft air samples with precisions of ∼1 %–2 % reported on the SIO-14 gravimetric calibration scale. Combined with inverse methods, we found near-zero atmospheric abundances from the early 1900s to the early 1960s, after which they rose sharply, reaching 1.66 ppt (parts per trillion dry-air mole fraction) in 2017. Global c-C4F8 emissions rose from near zero in the 1960s to 1.2±0.1 (1σ) Gg yr−1 in the late 1970s to late 1980s, then declined to 0.77±0.03 Gg yr−1 in the mid-1990s to early 2000s, followed by a rise since the early 2000s to 2.20±0.05 Gg yr−1 in 2017. These emissions are significantly larger than inventory-based emission estimates. Estimated emissions from eastern Asia rose from 0.36 Gg yr−1 in 2010 to 0.73 Gg yr−1 in 2016 and 2017, 31 % of global emissions, mostly from eastern China. We estimate emissions of 0.14 Gg yr−1 from northern and central India in 2016 and find evidence for significant emissions from Russia. In contrast, recent emissions from northwestern Europe and Australia are estimated to be small (≤1 % each). We suggest that emissions from China, India, and Russia are likely related to production of polytetrafluoroethylene (PTFE, “Teflon”) and other fluoropolymers and fluorochemicals that are based on the pyrolysis of hydrochlorofluorocarbon HCFC-22 (CHClF2) in which c-C4F8 is a known by-product. The semiconductor sector, where c-C4F8 is used, is estimated to be a small source, at least in South Korea, Japan, Taiwan, and Europe. Without an obvious correlation with population density, incineration of waste-containing fluoropolymers is probably a minor source, and we find no evidence of emissions from electrolytic production of aluminum in Australia. While many possible emissive uses of c-C4F8 are known and though we cannot categorically exclude unknown sources, the start of significant emissions may well be related to the advent of commercial PTFE production in 1947. Process controls or abatement to reduce the c-C4F8 by-product were probably not in place in the early decades, explaining the increase in emissions in the 1960s and 1970s. With the advent of by-product reporting requirements to the United Nations Framework Convention on Climate Change (UNFCCC) in the 1990s, concern about climate change and product stewardship, abatement, and perhaps the collection of c-C4F8 by-product for use in the semiconductor industry where it can be easily abated, it is conceivable that emissions in developed countries were stabilized and then reduced, explaining the observed emission reduction in the 1980s and 1990s. Concurrently, production of PTFE in China began to increase rapidly. Without emission reduction requirements, it is plausible that global emissions today are dominated by China and other developing countries. We predict that c-C4F8 emissions will continue to rise and that c-C4F8 will become the second most important emitted PFC in terms of CO2-equivalent emissions within a year or two. The 2017 radiative forcing of c-C4F8 (0.52 mW m−2) is small but emissions of c-C4F8 and other PFCs, due to their very long atmospheric lifetimes, essentially permanently alter Earth's radiative budget and should be reduced. Significant emissions inferred outside of the investigated regions clearly show that observational capabilities and reporting requirements need to be improved to understand global and country-scale emissions of PFCs and other synthetic greenhouse gases and ozone-depleting substances.
- Article
(5526 KB) -
Supplement
(3719 KB) - BibTeX
- EndNote





The perfluorocarbon (PFC) perfluorocyclobutane (c-C4F8, PFC-318, octafluorocyclobutane, CAS 115-25-3) is a very long-lived and potent greenhouse gas (GHG) regulated under the Paris Agreement of the United Nations Framework Convention on Climate Change (UNFCCC). Ravishankara et al. (1993) concluded that the most important atmospheric loss process of c-C4F8 is Lyman-α photolysis resulting in an atmospheric lifetime of 3200 years. Later, Morris et al. (1995) argued that if reactions of c-C4F8 with electrons and positive ions in the mesosphere and aloft are irreversible, the lifetime could be reduced to 1400 years, which, on human timescales, is still essentially infinite. c-C4F8 has a radiative efficiency of 0.32 W m−2 ppb−1 (parts per billion) and, assuming a 3200-year lifetime, a global warming potential of 9540 on a 100-year timescale (GWP100) (Myhre et al., 2013; Engel et al., 2018). Due to the long lifetime and high radiative efficiency, emissions of c-C4F8 (and other perfluorinated compounds) essentially permanently alter the radiative budget of Earth (Victor and MacDonald, 1999).
Lovelock (1971) predicted the accumulation of c-C4F8 in the global atmosphere, but to the best of our knowledge, the earliest atmospheric measurements of c-C4F8 were presented in Sturges et al. (1995) and in the PhD theses of Travnicek (1998) and Oram (1999, discussed further below). Sturges et al. (2000) determined from one vertical balloon-borne profile in 1994 that c-C4F8 mole fractions declined from ∼1.1 ppt (parts per trillion) in the lower atmosphere of the Northern Hemisphere (NH) to ∼0.6 ppt in the stratosphere, while Harnisch (1999) reported that Sturges et al. (1995) had found 0.4 ppt in the troposphere decreasing to ∼0.1 ppt at 25 km in 1994, suggesting a revised calibration scale. Harnisch et al. (1998) and Harnisch (1999) estimated from this atmospheric gradient global emissions of 1–2 Gg yr−1 (kt yr−1, 1 t = 0.001 Gg). Travnicek (1998) reported ∼0.2 ppt in 1977 and ∼0.7 ppt in 1997 in the NH troposphere, from which Harnisch (2000) estimated average global emissions of 0.7 Gg yr−1. Despite differences in early measurements and emission estimates, perhaps due to different calibration scales and analytical methods, these studies were consistent with the accumulation of c-C4F8 in the global atmosphere.
Harnisch (1999, 2000) stated that c-C4F8 had limited economic relevance, with some use for plasma etching in the semiconductor industry, that c-C4F8 can be formed via dimerization of tetrafluoroethylene (TFE), and that thermal decomposition or combustion of polytetrafluoroethylene (PTFE) and other fluoropolymers (Morisaki, 1978) (during waste disposal) possibly led to the accumulation of atmospheric c-C4F8.
Today we have stronger evidence for c-C4F8 emissions from the semiconductor and microelectronics industry as it has been increasingly used since the 1990s for dry etching, chemical vapor deposition chamber cleaning, and as deposition gas (Bosch process). Compared to other fluorinated gases used for these processes, more selective etching, cost reduction in plasma cleaning, easier abatement, and hence potentially lower contribution to global warming have been cited as advantages of c-C4F8 (e.g., Sasaki et al., 1998; Christophorou and Olthoff, 2001; Raju et al., 2003; Kokkoris et al., 2008; and references therein). However, due to efficient abatement with modern emission controls (up to 90 %), today's c-C4F8 emissions from this industry could also be small (Zhihong et al., 2001).
Today we also have further evidence that the thermal decomposition of PTFE and other fluoropolymers can lead to the formation of c-C4F8, TFE, and hexafluoropropylene (HFP) (van der Walt et al., 2008; Bezuidenhoudt et al., 2017); the resultant c-C4F8 could therefore be emitted to the atmosphere.
One potentially major source of c-C4F8 that seems to have received too little attention is the production of TFE and HFP monomers, the building blocks for PTFE, fluorinated ethylene propylene (FEP, TFE/HFP copolymer), and other fluoropolymers, which involves pyrolysis of hydrochlorofluorocarbon 22 (HCFC-22, CHClF2) as c-C4F8, the dimer of TFE, is a by-product/intermediate of this process (Chinoy and Sunavala, 1987; Broyer et al., 1988; Gangal and Brothers, 2015). This reaction can be steered towards HFP or c-C4F8 by controlling the dimerization of TFE to c-C4F8 and the co-pyrolysis of c-C4F8 with TFE to HFP (Jianming, 2006). c-C4F8 could therefore be emitted during TFE–HFP–PTFE–FEP production if it is not abated or recovered, e.g., for use in the semiconductor industry or for pyrolysis with TFE to HFP at a later stage, perhaps at a different facility.
Several other, perhaps minor, emissive uses of c-C4F8 are also known (see Lewis, 1989; Chung and Bai, 2000; Harnisch, 2000; Christophorou and Olthoff, 2001; Kim et al., 2002; Liu et al., 2008; and reference therein), e.g., in foamed/sprayed foods, as a food packaging gas, in retinal detachment surgery, for contrast-enhanced ultrasound imaging, in radar systems, as a specialty refrigerant (e.g., in submarines where R-405A (43 % c-C4F8) can replace pure HCFC-22 and the chlorofluorocarbon CFC-12, CCl2F2), as an electrically insulating dielectric gas (e.g., in mixtures with sulfur hexafluoride, SF6), as a medium for polymerization reactions, in fire extinguishers, and perhaps as a geohydrological tracer (Kass, 1998). Several chemical reactions in which c-C4F8 is used to introduce –CF3 groups into organic molecules are known (https://scifinder.cas.org/, last access: 19 June 2019) as well as reactions leading to desirable products such as HFO-1234yf, a fourth-generation refrigerant used in newer mobile air conditions (MACs; see Supplement) or HFP, but also various other compounds. Production of c-C4F8 for these uses, via the pyrolysis of HCFC-22 or perhaps from 1,2-dichlorotetrafluoroethane (CFC-114) (Siegemund et al., 2016), may cause emissions as well. While the major atmospheric PFC tetrafluoromethane (CF4) as well as the minor PFCs hexafluoroethane (C2F6) and octafluoropropane (C3F8) are released during primary aluminum production (Holliday and Henry, 1959; Tabereaux, 1994; Fraser et al., 2013), no evidence for c-C4F8 emissions has been presented so far. Cai et al. (2018) presented evidence for negligible emissions of c-C4F8 from the similar electrolytic production of rare earth elements in China. There are no known natural sources of c-C4F8. In summary, there may be multiple c-C4F8 emission sources, but the extent and time evolutions of these various potential emission sources are unclear.
Saito et al. (2010) reported the first continuous, approximately 4-year-long, in situ measurement record of c-C4F8 at two stations in the NH, with mean baseline 2006–2009 mole fractions of ∼1.22 ppt at Cape Oshiishi (43.1∘ N, 145.3∘ E) and ∼1.33 ppt at Hateruma Island (24.1∘ N, 123.8∘ E) (NIES calibration scale). Saito et al. (2010) determined increase rates of 0.01–0.02 ppt yr−1 and global emissions of 0.6±0.2 Gg yr−1.
Oram et al. (2012) published the first multi-decade-long atmospheric record of c-C4F8 in the Southern Hemisphere (SH). They combined previous measurements of subsamples of the Cape Grim Air Archive (CGAA) for the SH with air dates prior to 1994 (from Oram, 1999, converted to a new, 19.6 % lower calibration scale with an estimated uncertainty of ≤7 %) with newer measurements of CGAA subsamples with air dates after 1994 and a change of analytical method after 2006. They found an increase in c-C4F8 at Cape Grim from 0.35 ppt in 1978 to ∼0.8 ppt in 1995 and 1.2 ppt in 2010, with a current increase rate of ∼0.03 ppt yr−1. They reported that global c-C4F8 emissions increased from ∼0.9 Gg yr−1 in the early 1980s to ∼1.7 Gg yr−1 in 1986 before declining to a minimum of ∼0.4 Gg yr−1 in 1993, after which they increased to ∼1.1 Gg yr−1 in 2006 and 2007 and may have stabilized. Oram et al. (2012) noted that the global emissions determined by Saito et al. (2010) were lower than their estimate and suggested that the underlying atmospheric rise rate measured by Saito et al. (2010) may be too small.
In summary, calibration differences between previous studies are significant, no multi-decadal c-C4F8 record for the NH has been published, and global emissions have not been reassessed since Oram et al. (2012). Therefore our primary goals have been to develop an independent gravimetric c-C4F8 calibration scale and to characterize the abundances of c-C4F8 with high precisions in both hemispheres in order to determine updated historic and recent global emissions. We present measurements of c-C4F8 with precisions of ∼1 %–2 % on the SIO-14 calibration scale (∼2 % accuracy) developed by the Scripps Institution of Oceanography (SIO) using instrumentation and calibration methods of the Advanced Global Atmospheric Gases Experiment (AGAGE) program (Prinn et al., 2018). We discuss historic atmospheric mole fractions of c-C4F8 based on measurements of the CGAA for the extratropical SH, archived air samples from various sources for the extratropical NH, continuous atmospheric measurements in both hemispheres at multiple remote AGAGE stations since mid-2010, combined with measurements of air extracted from firn from both hemispheres. Using our measurements and inverse modeling methods, we infer global c-C4F8 emissions since the beginning of the 20th century until 2017. To improve our understanding of prominent c-C4F8 sources and source regions, we investigate regional c-C4F8 emission strengths as observed by the global AGAGE network in eastern Asia, Europe, parts of Australia, and Russia and by an aircraft campaign over India. We also summarize and discuss available inventory based “bottom-up” emissions and compare them to the emissions we determined with our atmospheric-measurement-based “top-down” approach.
2.1 Instrumentation, data availability, and calibration
c-C4F8 and ∼40 other halogenated compounds were measured by AGAGE in 2 L air samples with the Medusa cryogenic pre-concentration systems with a gas chromatograph (GC, Agilent 6890) and quadrupole mass selective detector (MSD) (Miller et al., 2008; Prinn et al., 2018). Data from 12 in situ measurement sites and 14 Medusa instruments were used. At Monte Cimone, Italy, c-C4F8 was measured with a commercial adsorption–desorption system with a gas chromatograph and mass spectrometer (ADS–GC–MS) (Maione et al., 2013). Table 1 shows the availability of in situ, archived air (Sect. 2.2), firn air (Sect. 2.3), and aircraft air sample (Sect. 2.4) measurements with information for each site. For all measurements, each sample was alternated with a reference gas (Prinn et al., 2000; Miller et al., 2008), resulting in up to 12 fully calibrated samples per day (Medusa and ADS-GC–MS). The reference gases at each site were calibrated relative to parent standards at SIO.
Table 1Availability of c-C4F8 in situ, flask, firn, and aircraft air measurements, measurements sites, and instrumentation.
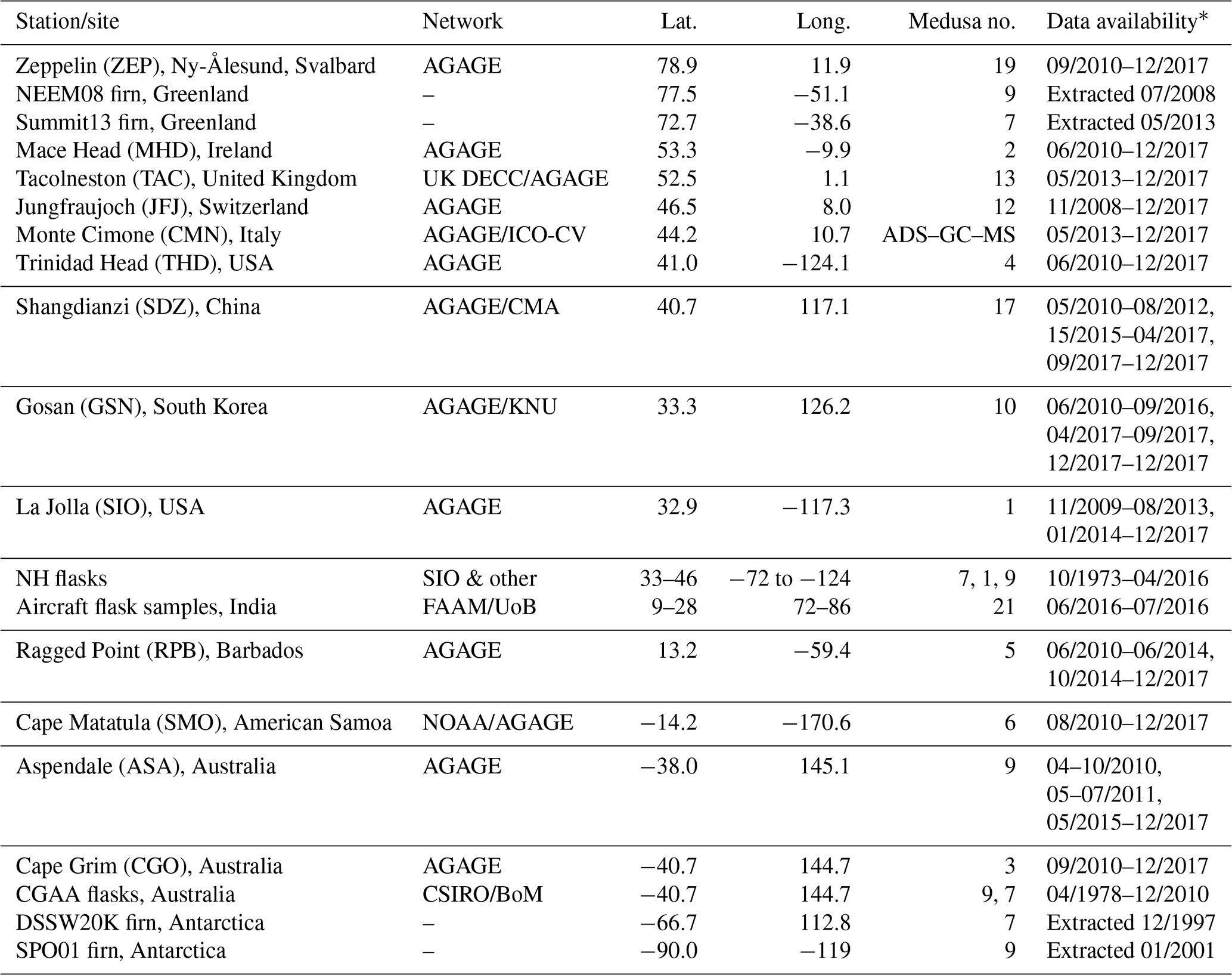
∗ Shorter interruptions are excluded. AGAGE: Advanced Global Atmospheric Gases Experiment (Prinn et al., 2018). NEEM08: firn air samples collected in 2008 at the Northern Greenland Eemian Ice Drilling Project, Greenland, were collected by the University of Copenhagen, Denmark, the NEEM consortium, and the Commonwealth Scientific and Industrial Research Organisation (CSIRO) (Buizert et al., 2012). Summit13: Firn samples collected in 2013 near Summit station, Greenland, by the University of Rochester and Oregon State University. UK DECC: the Tacolneston (TAC) site is part of the UK Deriving Emissions linked to Climate Change network (Stanley et al., 2018). DSSW20K: firn samples collected in December 1997 at Dome Summit South West 20 km, Law Dome, by CSIRO, the Australian Antarctic Division (AAD), and the Australian Nuclear Science and Technology Organisation (ANSTO) (see Trudinger et al., 2016, and citations therein). SPO01: firn samples collected in 2001 at South Pole, Antarctica, by Bowdoin College, the National Oceanic and Atmospheric Administration (NOAA), the University of Colorado and the National Science Foundation (NSF) (Aydin et al., 2004; Sowers et al., 2005). ICO-OV: measurements at the Italian Climate Observatory “O. Vittori” Monte Cimone (CMN) were performed with a commercial adsorption–desorption system with a gas chromatograph and mass spectrometer (ADS–GC–MS) (Maione et al., 2013). CMA: China Meteorological Administration. KNU: Kyungpook National University, South Korea. SIO & other: most archived northern hemispheric (NH) samples were collected by the Scripps Institution of Oceanography, La Jolla, and measured on Medusa 7. FAAM/UoB: air samples over India and the Indian Ocean were taken aboard the UK's FAAM (Facility for Airborne Atmospheric Measurements) BAe-146 research aircraft and analyzed on Medusa 21 at the University of Bristol (UoB) (Say et al., 2019). CGAA: Cape Grim Air Archive samples were collected by the CSIRO Oceans and Atmosphere and the Bureau of Meteorology (BoM), Australia, and predominantly measured on the Aspendale Medusa 9 at CSIRO (Langenfelds et al., 2014; Fraser et al., 2018).
c-C4F8 measurements are reported on the SIO-14 calibration scale as part per trillion dry-air mole fractions. The calibration scale is based on four gravimetric halocarbon∕nitrous oxide (N2O) mixtures via a stepwise dilution technique with large dilution factors for each step (103 to 105) (Prinn et al., 2000, 2001). High-purity c-C4F8 (99.999 %, Matheson Tri-gas) and N2O (99.9997 %, Scott Specialty Gases) were further purified by repeated cycles of freezing (−196 ∘C), vacuum removal of non-condensable gases, and thawing. Artificial air (ultra-zero grade, Airgas) was further purified via an absorbent trap filled with glass beads, molecular sieve (MS) 13X, charcoal, MS 5Å, and Carboxen 1000 at −80 ∘C (ethanol/dry ice). Zero air was measured to verify insignificant c-C4F8 and other halocarbon blank levels before being spiked with the c-C4F8∕N2O mixtures. The resulting mixtures of c-C4F8 in artificial air have prepared values of ∼1.3 ppt and the relative standard deviation of the calibration scale is 0.23 %. We estimate the uncertainties of the calibration scale propagation from SIO to the sites to be ∼0.6 % and the calibration scale uncertainty to be ∼2 % (see Prinn et al., 2000, 2001, 2018).
The primary calibration instrument for the AGAGE network at SIO (La Jolla, California), Medusa 1, and all field instruments used a Porabond Q (25 m, 0.32 mm I.D., 5 µm film thickness, Varian) chromatographic main column and, initially Agilent 5973, later 5975 series MSDs. The original Medusa design is described by Miller et al. (2008); subsequently all Medusas were converted or newly built to measure nitrogen trifluoride (NF3) (Arnold et al., 2012), but this did not affect the c-C4F8 measurement methodology or the results. While 5975 MSDs are beneficial for samples and compounds with very low mole fractions, precisions for c-C4F8 measurements of archived air samples (3–7 replicates, see next section) were similar, i.e., better than ∼0.01 ppt. Daily reference gas measurement precisions slightly improved from ∼0.02 ppt (∼1.5 %–2 %) to ∼0.01 ppt (∼1 %–1.5 %) with the 5975 MSDs. Detection limits (3 times baseline noise) for 2 L air samples were ∼0.01–0.03 ppt for both types of MSDs.
In addition to calibrations, Medusa 1 was also used to measure in situ local ambient air and several archived air samples (see Sect. 2.2). However, analysis of most archived air samples at SIO occurred on a second instrument, Medusa 7, as it was equipped with a more sensitive 5975 MSD at that time. For these measurements, we temporarily converted Medusa 7 to use a GasPro GSC (60 m, 0.32 mm I.D., Agilent) main column as it promised better separation performance for several higher PFCs (Ivy et al., 2012) measured along with c-C4F8. Similarly, Medusa 9, the instrument used to measure most CGAA samples at the Commonwealth Scientific and Industrial Research Organisation (CSIRO, Aspendale) and ambient air after October 2010, had been converted to use a GasPro column. On both types of main columns, c-C4F8 was measured on mass-over-charge ratios (m∕z) of 131 () and 100 () and reported by height using carefully chosen integration parameters as perfluorobutane (C4F10) shares both m∕z and elutes on the tail of c-C4F8. The m∕z ratios remained the same despite the very different separation principles of these two main columns. Measurements of archived air samples on Medusa 7 with both main columns agreed within less than 0.01 ppt (ratio of 1.0016, R2=1.0000, n=4, 0.237–1.11 ppt). In situ c-C4F8 measurements at SIO with Medusa 1 (Porabond Q) and 7 (with the GasPro column) continued to agree within typical precisions. We also compared archived air measurements on Medusa 1 and 7, both before and while Medusa 7 used the GasPro column, and results agree within precisions of 0.02 ppt or better (Medusa 1 vs. Medusa 7, both Porabond Q, ratio of 1.0001, R2=0.9987, n=95, 0.237–1.616 ppt, Medusa 1, Porabond Q vs. Medusa 7, GasPro, ratio of 1.0018, R2=0.9979, n=39, 0.239–1.515 ppt). These tests show that the different main columns did not cause any bias.
The analytical systems showed no significant c-C4F8 blanks. The linearity of Medusa 7 (SIO) and 9 (CSIRO) used to measure archived air samples was assessed with a series of diluted air samples (parent tank at 1.252 ppt, dilutions from 100 % to 6.25 %; Ivy et al., 2012) and a series of different volumes of a working standard (parent tank at 1.60 ppt, sample volumes from 200 % to 5 % of usual 2 L volume). A small deviation from linearity was observed for the most diluted samples and the smallest volumes, probably due to a memory or blank of ∼0.014 ppt on Medusa 9, for which a correction was applied. Medusa 7 showed an effect of ∼0.008 ppt, but as this was just below the detection limits and within the typical precisions, we chose not to correct for this.
2.2 Archived air samples of the extratropical Southern Hemisphere (SH, Cape Grim Air Archive, CGAA) and extratropical Northern Hemisphere (NH)
To reconstruct the atmospheric history of c-C4F8 in the extratropical SH, 41 unique CGAA samples (collected 1978–2009; Langenfelds et al., 2014) were measured at CSIRO in 2011 (Ivy et al., 2012). In addition, eight subsamples of CGAA parent tanks and four additional SH samples were measured at SIO to demonstrate that measurements at CSIRO and SIO agree (for details see the Supplement). Based on an iterative filtering process designed to reject outliers greater than 2σ deviations from curve fits through the results for all 60 SH samples (41 at CSIRO and 19 at SIO) and pollution-filtered monthly mean measurements (O'Doherty et al., 2001; Cunnold et al., 2002) at the extratropical stations CGO and ASA (Australia), 13 SH samples were rejected as outliers, leaving 47 SH samples (78 %).
To reconstruct the atmospheric history in the extratropical NH, 126 unique air samples mostly filled at SIO and THD (1973–2016) were measured at SIO. Additionally, three NH samples (filled in 1980 and 1999) were measured at CSIRO to demonstrate that measurements at CSIRO and SIO agree (for details see the Supplement). Most of the NH samples had been filled during baseline conditions for various purposes using modified diving compressors (RIX Industries, US, SA-3 and SA-6, Weiss and Keeling laboratories) and did not show any artifacts for many gases (e.g., Mühle et al., 2010; O'Doherty et al., 2014; Vollmer et al., 2016). For c-C4F8, however, comparisons with concurrent in situ measurements at MHD, THD, SIO, and JFJ revealed artifacts for most of these samples and the iterative filtering process only retained c-C4F8 data for 11 NH samples. In contrast, CGAA tanks were filled with a cryogenic method which did not produce any bias. Due to the sparse NH data and poor data quality before in situ measurements started in the NH, the fits used for the iterative filtering process of NH data had to be guided by the final SH fit shifted by 1.5 years to allow for the delay of c-C4F8 accumulation between the SH and NH due to interhemispheric transport (Mühle et al., 2010; Vollmer et al., 2016). Without this guidance, initial NH fits were dominated by high outliers, resulting in bad fits. It should be pointed out that most of the filtered NH tanks were filled in 2003 and later, typically many tanks on one or two days in a given year, which would add little information to the reconstruction given the onset of in situ data at multiple stations in 2011 and the high quality of the CGAA data used to guide the filtering. Figure 1 shows the filtered data and the final suggested fits and 95 % confidence bands.
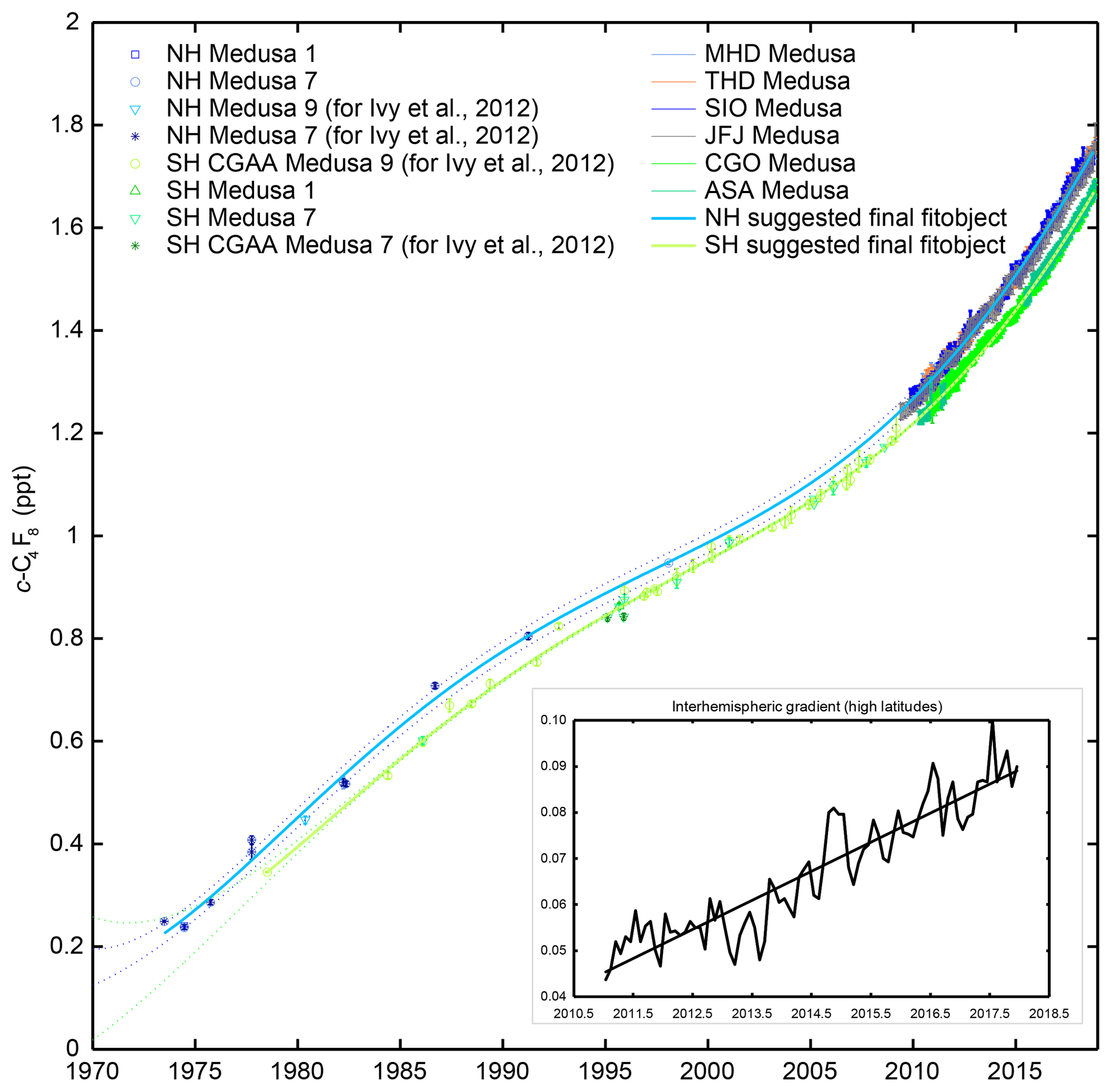
Figure 1c-C4F8 mole fractions reconstructed from the late 1970s to 2018 from archived air samples and in situ measurements in both hemispheres. Cape Grim Air Archive (CGAA) and archived NH air samples are shown with symbols in shades of green and blue, respectively, reflecting different data subsets. For recent years, in situ measurements are shown as pollution removed monthly means for extratropical stations in the NH (MHD in light blue, THD in orange, SIO in darker blue, JFJ in grey) and in the SH (CGO in lighter green, ASA in pale green). Shown are the final data after an iterative filtering process described in the main text. The final suggested fits are shown as bold light green (SH) and bold light blue (NH) polynomial fits. Confidence bands (2σ) are shown as dotted lines. Results for the tropical stations, RPB and SMO, the Asian stations, GSN and SDZ, and the Arctic station, ZEP, are omitted here for clarity. For individual samples, error bars reflect measurement precisions. For monthly means, error bars represent standard deviations. The inset shows the interhemispheric gradient from in situ measurements at high latitudes (MHD, THD, SIO, and CGO) from 2011 to 2017.
2.3 Air extracted from firn
To augment the data set of in situ and archived air measurements, we measured c-C4F8 in samples from a subset of the firn sites described in Trudinger et al. (2016), namely NEEM08 in the NH and DSSW20K and SPO01 in the SH, plus one new site in the NH, Summit13, Greenland. We used the CSIRO firn model (Trudinger et al., 1997, 2013) to characterize the age of the air in these samples (detailed in Sect. 4.1). Here, we give a brief description of the firn sites. For a full description of the calibration of the CSIRO firn model for NEEM08, DSSW20K, and SPO01, see Trudinger et al. (2013), and for Summit13 see Fig. S1 in the Supplement.
NEEM08. Firn air was extracted from the EU borehole in July 2008 in northern Greenland, drilled near the North Greenland Eemian Ice Drilling Project (NEEM) deep ice core drilling site (77.45∘ N, 51.06∘ W) (Buizert et al., 2012). This site has a moderate snow accumulation rate of 199 kg m−2 yr−1.
Summit13. Firn air was collected in May 2013 at Summit, Greenland, from a borehole (72.66∘ N, 38.58∘ W) drilled 10 km NNW of Summit Station, Greenland. The US Firn Air system (Battle et al., 1996) was used to extract the air from 19 depth levels in the firn from the surface to just above 80.06 m (below this depth firn air can no longer be collected as the open channels in firn have closed off and formed discrete bubbles embedded in ice). The 3 in. borehole was drilled with the Eclipse Ice Drill (IDDO) and new rubber bladders (1∕8 in. thick) were fabricated (Greene Rubber Co., Woburn, MA) for use in this campaign. The 2.5 L glass flasks were filled at all depths for high-resolution measurements of gases performed by the National Oceanic and Atmospheric Administration (NOAA) (CO2, CH4, CO, N2O, SF6, H2). Larger volume samples from preselected depth levels were filled in 35 L electro-polished SS tanks using a KNF Neuberger pump (with neoprene diaphragms). These samples were measured at SIO for c-C4F8 and other trace gases (including CH4, N2O, CFCs, HFCs, HCFCs, and SF6). For quality control purposes, the sample line was measured on site for CO2 and CH4 by cavity ring-down spectroscopy (CRDS, Los Gatos Research, μ-GGA) and CO by a reducing compound photometer (Peak Labs, RCP1) prior to filling the flasks. Summit has a moderate snow accumulation rate of 211 kg m−2 yr−1. CSIRO firn model calculations for Summit use the density profile from Adolph and Albert (2014) and mean annual temperature and pressure of 241.75 K and 665 mbar. The diffusivity profile and related parameters were calibrated using the measurements of CO2, CH4, N2O, SF6, CFC-11, CFC-12, CFC-113, CH3CCl3, HFC-134a, HCFC-141b, and HCFC-142b described above. Firn model results for these tracers are shown in Fig. S1.
DSSW20K. Firn air was collected in January 1998 in eastern Antarctica (66.73∘ S, 112.83∘ E) from a borehole drilled 20 km west of the deep Dome Summit South (DSS) drill site near the summit of Law Dome (Smith et al., 2000; Sturrock et al., 2002; Trudinger et al., 2002). This site has a short firn column and a moderate snow accumulation rate of 150 kg m−2 yr−1.
SPO01. We only measured one sample collected in 2001 from 120 m from a borehole at the South Pole, Antarctica (90∘ S, 119∘ W) (Aydin et al., 2004; Sowers et al., 2005). This site has a deep firn column and a low snow accumulation rate of 75 kg m−2 yr−1, resulting in old firn air.
Firn air extracted from the DSSW20K, NEEM08, and SPO01 sites was measured at CSIRO in 2012 (Medusa 9), while Summit13 firn air was measured at SIO (Medusa 7), see Table 1. c-C4F8 firn measurement data are included in the data file listed in the Supplement. Other gases such as CH4 and N2O were measured as well.
2.4 Air samples collected over India and the Indian Ocean
Air samples were collected on board the UK FAAM (Facility for Airborne Atmospheric Measurements) BAe-146 aircraft during 11 flights conducted from 12 June to 9 July 2016 (9–28∘ N, 72–86∘ E) into 3 L pre-evacuated electropolished stainless steel flasks (SilcoCan, Restek) sealed with metal bellows valves (SS-BNVVCR-4, Swagelok). During the time it took to compress the air samples to 3.8 bar (30–60 s, depending on altitude) using a metal bellows pump (PWSC 28823-7, Senior Aerospace, USA), the aircraft traveled ∼7 km. Nine flights occurred over northern India and two over southern India and the Indian Ocean. In total, 176 flask samples were collected, with the majority (> 90 %) of these samples filled below 1.5 km altitude. The size of the subsamples analyzed with Medusa 21 at the University of Bristol was reduced to 1.75 L (from 2 L) and the sampling rate to 50 mL min−1 (from 100 mL min−1) to allow for triplicate analyses of each flask and to accommodate for the lower flask pressure. c-C4F8 measurements are reported on the SIO-14 calibration scale. Detection limits, blanks, and precisions were similar to those stated above. For further details, see Say et al. (2019).
Emissions of compounds, such as c-C4F8, into the atmosphere are often estimated by so called “bottom-up” methods, which are based on information such as purchased, produced or imported amounts, industrial activities referred to as activity data, and estimated emission factors for each emissive process. Developed countries report annual emissions of GHG, including c-C4F8, to the UNFCCC using such bottom-up methods. However, these data are inherently not representative of total global emissions since developing countries do not have the same comprehensive UNFCCC reporting requirements, including countries such as South Korea, China, and Taiwan with sizable electronics and PTFE manufacturing capacities and thus with potentially significant c-C4F8 emissions. An additional complication is that several countries report unspecified mixes of PFCs or of PFCs and HFCs and other fluorinated compounds, making it difficult or impossible to estimate emissions of individual compounds, such as c-C4F8. In the Supplement, we gather available inventory information from submissions to UNFCCC, National Inventory Reports (NIRs), the Emissions Database for Global Atmospheric Research (EDGAR), the World Semiconductor Council (WSC), and the U.S. Environmental Protection Agency (EPA) in an effort to estimate contributions from unspecified mixes and countries not reporting to UNFCCC to compile a meaningful bottom-up inventory. Globally these add up to 10–30 t yr−1 (0.01–0.03 Gg yr−1) from 1990 to 1999, 30–40 t yr−1 (0.03–0.04 Gg yr−1) from 2000 to 2010, and 100–116 t yr−1 (∼0.1 Gg yr−1) from 2011 to 2014 (with a substantial fraction due to the US emissions from fluorocarbon production reported by the U.S. EPA). As has been found by Saito et al. (2010) and Oram et al. (2012), we show in Sect. 5.2 and 5.3 that measurement-based (top-down) global and most regional emissions are significantly larger than the compiled bottom-up c-C4F8 emission inventory information (see Fig. 5), analogous to what has been found for other PFCs (Mühle et al., 2010), reflecting the shortcomings of current emission reporting requirements and inventories.
4.1 CSIRO firn model
The CSIRO firn model and its use in global inversion frameworks has been described in detail (Trudinger et al., 2013, 2016; Vollmer et al., 2016, 2018, 2019). Air samples taken far away from pollution sources represent the background atmospheric trace gas composition at that time. Once air enters the firn, vertical diffusion and other physical processes in the firn lead to mixing of air of different ages. Therefore, air extracted from firn must be described with an age distribution. We used the CSIRO firn model to describe the relationship between trace gas mole fractions measured in each extracted air sample from a given depth and the corresponding age distribution of high-latitude atmospheric mole fractions. The diffusion coefficient of c-C4F8 relative to that of CO2 in air at 253 K used here was 0.47 with an estimated uncertainty of ∼10 %. This value was determined using Eq. (4) from Fuller et al. (1966) with Le Bas volume increments (e.g., Table 1.3.1, Mackay et al., 2006 and a multiplier for the Le Bas increments of 0.97, which minimizes the difference of calculated relative diffusion coefficients of a number of compounds from values measured by Matsunaga et al., 1993, 2002, 2005).
Figure 2 shows the measured depth profile of c-C4F8 (ppt) in air extracted from polar firn sites in the NH (Greenland) and the SH (Antarctica); for site details see Table 1. All samples showed c-C4F8 mole fractions above the detection limit. The firn reconstructed depth profiles are discussed in Sect. 4.3.1.
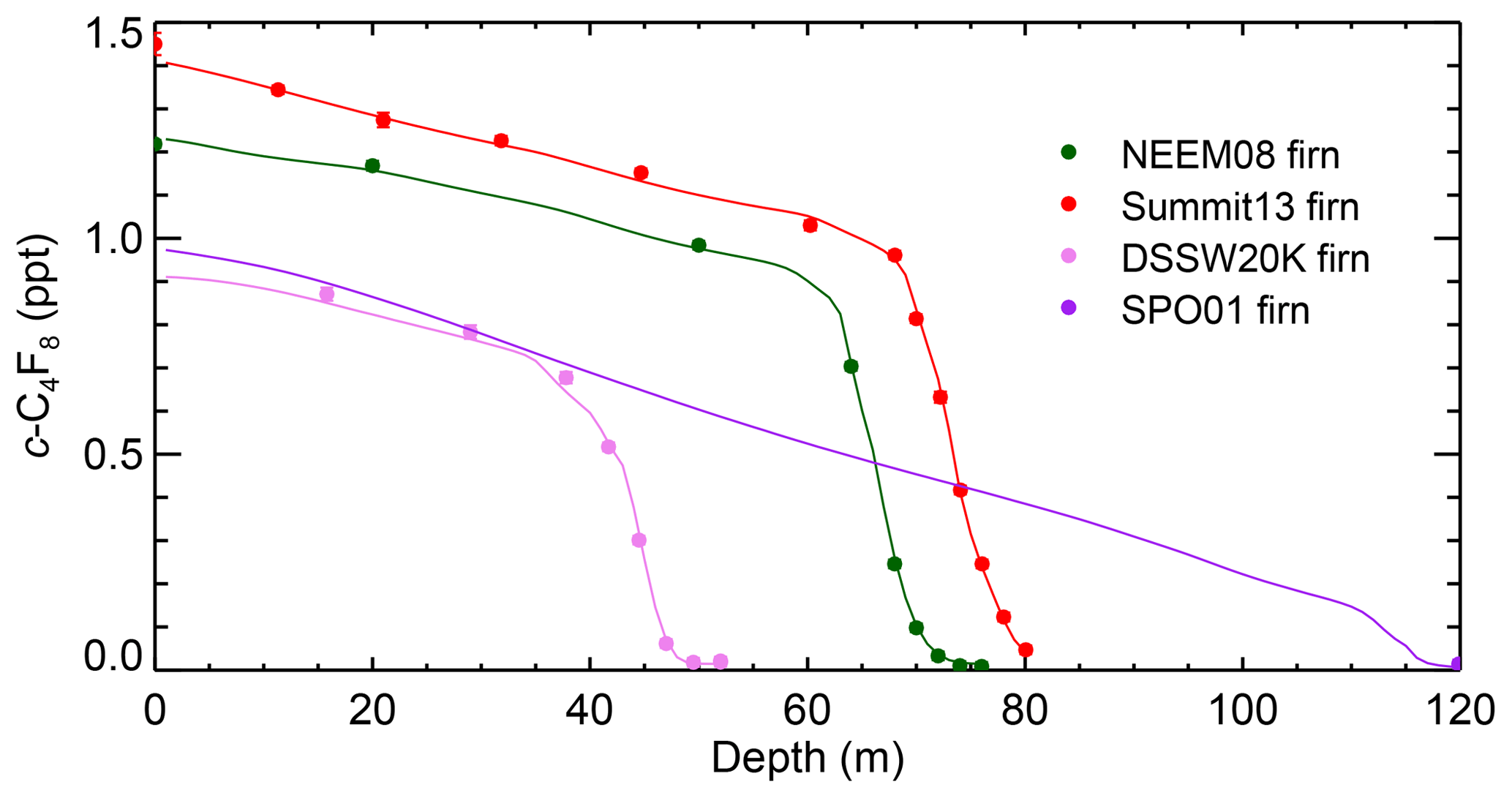
Figure 2Depth profile of c-C4F8 measured dry-air molar mole fractions (parts per trillion, ppt) in air extracted from polar firn at NEEM08 (northern Greenland, dark green) and Summit13 (Greenland, red) in the NH and DSSW20K (eastern Antarctica, pink) and SPO01 (South Pole, purple) in the SH, together with the simulated depth profiles for each site (dark green, red, pink, and purple lines) that correspond to the emissions inferred by the CSIRO inversion. The modeled depth profiles for each site (solid lines) are based on the inversion of measurements from all firn sites, archive, and in situ data. Measurement precisions (1σ) are shown as error bars and are generally smaller than the plotting symbol.
4.2 AGAGE 12-box model of the global atmosphere
The AGAGE 12-box two-dimensional model (Cunnold et al., 1983, 1997; Rigby et al., 2013) describes the transport and loss of trace gases in the global atmosphere. The model divides the atmosphere into four latitudinal bands at 0∘ and 30∘ S and ∘ N and three altitude bands at 500 and 200 hPa and calculates the mole fractions in each box. The AGAGE background sites (MHD, THD, RPB, SMO, and CGO; see Table 1) were historically chosen to represent the trace gas mole fractions in the four lower (tropospheric) model “boxes”. Model transport parameters were varied seasonally, but repeated annually. Given the very long atmospheric lifetime of c-C4F8 compared to the study period, the lifetime of c-C4F8 was assumed to be infinite in the model.
4.3 Global inversion methods
We used the AGAGE 12-box model in two different Bayesian inversions, denoted as the “CSIRO” and “Bristol” inversions, to estimate historic c-C4F8 emissions from our observations and to reconstruct historic abundances. Both inversions used in situ and archive data and the CSIRO inversion additionally used firn data. The observations need to be representative of clean background air at each sampling location. For in situ data, the AGAGE statistical method was used to remove pollution events and to calculate pollution-free monthly mean background air mole fractions for each AGAGE station (O'Doherty et al., 2001; Cunnold et al., 2002). As explained in Sect. 2.2, an iterative filtering algorithm starting out with all the archived air data and the pollution-free monthly means was then used to reject outliers for the extratropical SH and NH, mostly from the NH archive data. Due to the remoteness of the firn sample sites, we assumed background conditions without any filtering.
4.3.1 CSIRO inversion
The CSIRO inversion was developed to infer annual emissions at the global scale from firn, ice core, and atmospheric measurements (Sturrock et al., 2002; Trudinger et al., 2002, 2016). Green's functions from the CSIRO firn model were used to relate the measured air in the firn samples to air in the atmosphere in the past, and Green's functions from the AGAGE 12-box model were used to relate global emissions with a specified latitudinal distribution to mole fraction in the extratropical SH and NH. The inversion included constraints to avoid negative mole fractions, negative emissions, and unrealistic changes in emissions; these constraints were required due to the characteristics of inverting firn data and sparse archive data. The uncertainties in reconstructed mole fractions and inferred emissions were calculated using a bootstrap method that included the uncertainty in firn measurements, annual mean mole fraction (this uncertainty is temporally correlated; see Supplement in Vollmer et al., 2019), calibration scale (±2 %), and the firn model through the use of an ensemble of Green's functions corresponding to different firn model parameters including relative diffusivity (Trudinger et al., 2013, 2016; Vollmer et al., 2016).
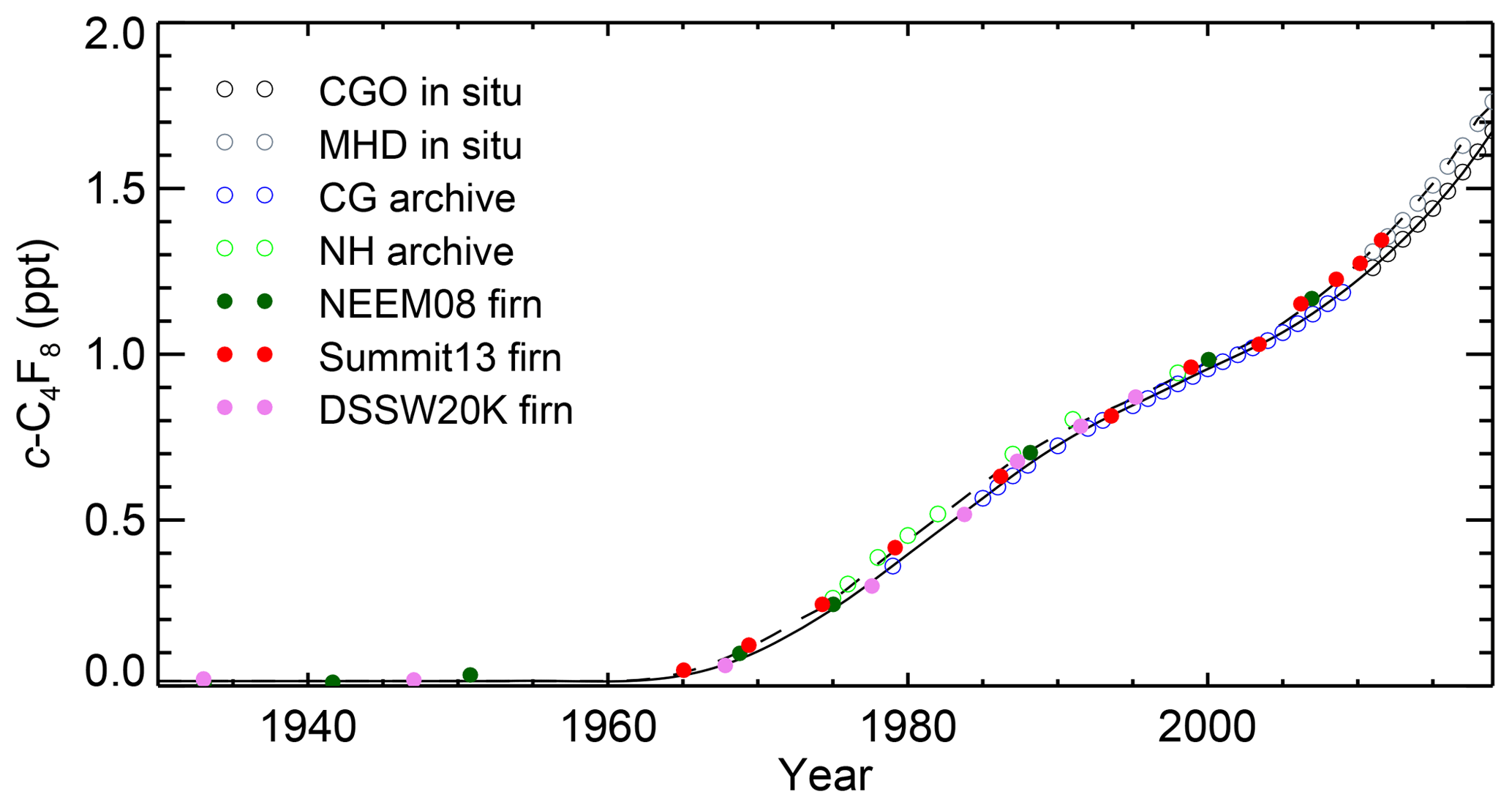
Figure 3Historic atmospheric c-C4F8 mole fractions reconstructed for the extratropical Northern Hemisphere and Southern Hemisphere from air extracted from polar firn (full circles, NEEM08 in dark green, Summit13 in red, DSSW20K in pink, against mean or effective ages; SPO01 with mean age of ∼1890 is not shown), annual values from spline fits to Cape Grim Air Archive (CG archive, open blue circles) and in situ measurements at Cape Grim (CGO, open black circles), archived air samples (NH archive, open green circles), and in situ measurements at Mace Head (MHD, open grey circles). Also shown are reconstructed abundances based on optimized emissions determined by the CSIRO inversion for the extratropical SH (black line) and NH (dashed black line).
Figure 3 shows the data that were used in the CSIRO inversion: annual values based on 10-year smoothing spline fits (i.e., 50 % attenuation at periods of 10 years) to monthly means of pollution-free in situ measurements at the AGAGE background sites CGO (SH) and MHD (NH), annual values based on 10-year smoothing spline fits to measurements of the CGAA and archived NH air samples, and air extracted from polar firn in both hemispheres. Annual means from the spline were only used in the inversion when there were pollution-free archive or in situ measurements around that time. Figure 3 also shows the final reconstructed abundances for the extratropical SH (solid black line) and NH (dashed black line) based on the optimized emissions. The measured mole fractions in firn air are plotted against their effective atmospheric ages if that age is after 1965, where the effective ages are calculated using the reconstructed history of atmospheric mole fractions determined by the CSIRO inversion (Trudinger et al., 2002). Before 1965, the growth rate in the atmosphere was small and uncertain; this makes it difficult to determine effective ages, so the earlier firn measurements are plotted against their mean ages (see also Fig. S7). Firn depth profiles for each firn site corresponding to the CSIRO inversion results are shown in Fig. 2 (solid lines) and they typically agree with the measurements within precisions (1σ, shown as error bars).
Overall, the abundances reconstructed with the CSIRO inversion agree very well with the measurement data (see also Fig. S2). In Fig. S3, we show the effect of excluding different sites from the inversion on reconstructed emissions and mixing ratios and the sensitivity of the inversion to the relative diffusion coefficient of c-C4F8.
It should be pointed out that the deepest NEEM08 firn air sample for the NH showed slightly lower mole fractions (0.0085 ppt) than the deepest DSSW20K samples for the SH (0.021 and 0.0185 ppt), although the mean ages are similar (1930s). The same applies to the second deepest NEEM08 (0.0105 ppt) and DSSW20K (0.018 ppt) samples (1940s), which is unexpected for a long-lived anthropogenic compound predominantly emitted in the NH. While the differences seem significant within the nominal precisions (0–0.0014 ppt) achieved for these firn samples measured only one to two times, they are not significant within typical precisions achieved for archive samples (∼0.01–0.02 ppt), which are typically measured three or more times and these data are just at or below the typical detection limits of 0.01–0.03 ppt. Based on the order in which the firn samples were measured and the absence of detectable blanks, it seems unlikely that a small blank, memory, calibration, or measurement problem could have caused this small discrepancy. The early part of the reconstructed record, with near-zero mole fractions, is also most susceptible to small uncertainties in the calibrated diffusivity profiles versus depth for all sites used in the firn model, uncertainties in the firn model structure (e.g., physical properties being invariant of time), or uncertainties in the diffusivity of different tracers relative to each other. Thus, there are a number of possible reasons for the higher mixing ratio in the SH firn data at this time, and we do not interpret this as evidence of higher mole fraction in the SH in the 1930s or 1950s.
4.3.2 Bristol inversion
The Bristol inversion was used to estimate annual fluxes of c-C4F8 using archive and in situ observations only (Rigby et al., 2011, 2014; Vollmer et al., 2018). A priori, it was assumed that emissions were similar from year to year such that the a priori year-to-year emission growth rate was assumed to be zero with an uncertainty of 200 t yr−2 (0.2 Gg yr−2, 1σ), approximately twice the bottom-up estimate in Sect. 3. The derived emission uncertainties include contributions from the measurement repeatability, the calibration scale uncertainty, and the model–measurement representation error (Rigby et al., 2014). Furthermore, because some archive air samples exhibit substantial short-timescale (< 1 year) variations that are unlikely to represent real changes in the background atmosphere (Fig. 11), the minimum uncertainty was set to the maximum deviation of the archive air samples from the smooth curve in Fig. 11 (0.03 ppt). Model representation errors were estimated as the variability of the pollution-free monthly baseline means determined by the AGAGE pollution algorithm (O'Doherty et al., 2001; Cunnold et al., 2002) from the high-frequency in situ data at each station for each given month. For periods without in situ data, the representation error was assumed to be equal to the average baseline variability from in situ data in the same latitudinal band scaled by the measured c-C4F8 abundance. The calibration scale propagation uncertainty is estimated based on propagation uncertainties of the c-C4F8 calibration scale from primary gravimetric standards to secondary standards within the “R1” relative calibration framework used in AGAGE and on propagation uncertainties from the R1 framework to the standards used to measure individual samples. Figure 4 shows that there is good agreement between the archived air samples (Sect. 2.2) and the pollution-free monthly mean in situ data from the AGAGE background sites (MHD and THD, RPB, SMO, and CGO) used in the Bristol inversion and the reconstructed mole fractions for the four latitudinal bands which these samples represent (see also Fig. S4).
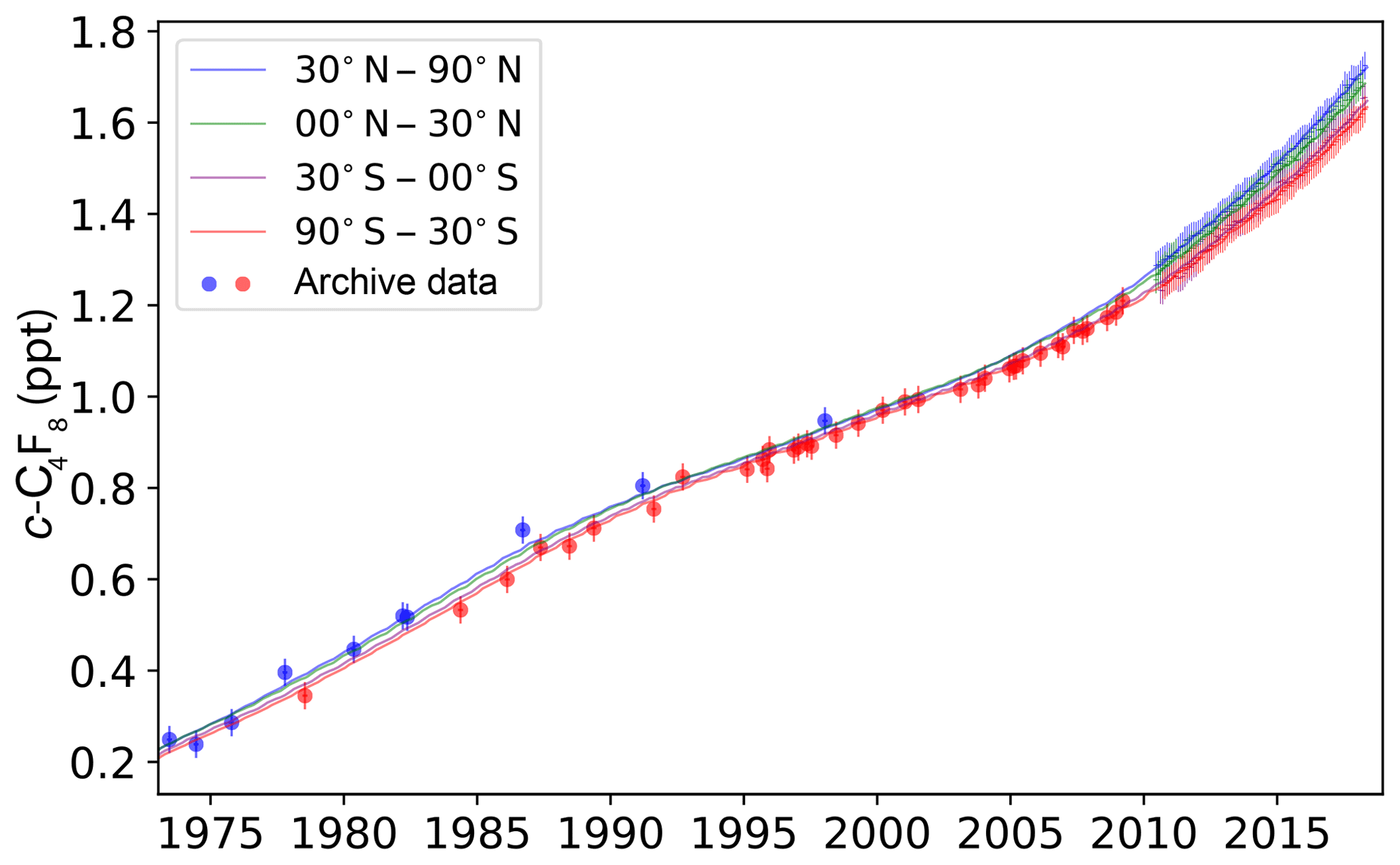
Figure 4Historic c-C4F8 mole fractions from archive samples in both hemispheres (filled circles) and pollution-free monthly mean in situ data from AGAGE background sites (MHD and THD in blue, RPB in green, SMO in purple, and CGO in green vertical bars; bar size represents variability of monthly means) are shown together with the Bristol inversion results for the four latitudinal bands represented by these background sites (30–90, 0–30∘ N, 0–30, and 30–90∘ S, solid lines of same color).
4.4 Regional model and inversion study using NAME-HB for eastern Asia
To investigate regional emissions in eastern Asia (20–50∘ N and 110–160∘ E) from our observations we used an inversion method based on Bayesian inference. We estimated annual mean emissions, assuming that emissions are constant in both space and magnitude during each calendar year. Here, the inversion used observations from the Gosan station as this site was operated with relatively few interruptions from October 2010 to the end of 2017, with best data coverage from 2011 to 2015. These observations were binned into 12 hourly averages. The inversion method requires an atmospheric transport model to derive the sensitivity of the observations to a surface emission field. We used the Lagrangian NAME (Numerical Atmospheric dispersion Modelling Environment) model from the UK Met Office (Jones et al., 2007), driven by meteorology from the Met Office Unified Model (Walters et al., 2014). The sensitivity was derived by releasing 20 000 hypothetical air parcels per hour of measurement from Gosan station, which were transported backwards in time for up to 30 d. The model recorded the time and location that air parcels interacted with the surface (below 40 m above ground level at a spatial resolution of 0.352∘ by 0.234∘), and these data were used to form an aggregated 30 d sensitivity or “footprint” map for each hour of measurement. In addition, the model recorded the time and location that air parcels left the domain boundaries to provide the sensitivity to the boundary conditions. The footprint maps, generated over the domain 5∘ S–74∘ N and 55–192∘ E and up to 19 km, were aggregated into 12 h averages.
We used a trans-dimensional hierarchical Bayesian method (NAME-HB) with a Metropolis–Hastings Markov chain Monte Carlo (MCMC) algorithm (Metropolis et al., 1953; Hastings, 1970) to solve the inverse problem. This allowed spatial emission estimates of c-C4F8 to be derived, whilst considering the uncertainties in the model, measurements, and a priori information and, importantly, the uncertainty in these uncertainties. Bayesian methods require a priori knowledge, here the emissions and boundary conditions. As little information on eastern Asia's c-C4F8 emissions (see Sect. 3) was available, we based our mean a priori emissions on those estimated by Saito et al. (2010). We spread their emissions for each reported country uniformly over the area of each country, rather than use population density (as in Saito et al., 2010) as that is not likely a good proxy of c-C4F8 emissions. We also spread 0.11 Gg yr−1 of emissions over the rest of the domain where the footprint was calculated. The value of 0.11 Gg yr−1 is an approximate scaling of the global total emissions based on population in this outer domain, i.e., the remainder of the domain not defined as eastern Asia. While we do not report emission estimates outside of eastern Asia due to large posterior uncertainties, they are still estimated in the inversion as they are useful when modeling the emissions in eastern Asia and their uncertainties that we do report. We assigned a large uncertainty to these a priori emissions (Table S1), which were governed by a lognormal distribution, so that they were uninformative and the observations dominated the estimation. We set a priori boundary conditions to be the mean background mole fractions measured at MHD on each vertical boundary (N, E, W, S) of the NAME domain. Offsets to the boundary conditions on each boundary were estimated in the inversion on a monthly basis.
The hierarchical nature of the inversion method means that hyper-parameters were also incorporated to include uncertainties in the NAME sensitivities, which are described by a multivariate normal distribution (see Ganesan et al., 2014). The reversible jump, or trans-dimensional, aspect of the inversion means that the underlying resolution at which the emissions are estimated is itself explored during inference (Lunt et al., 2016). Table S1 shows the a priori probability distributions assigned to the emissions and boundary condition scaling factors, model uncertainty, and the underlying grid. The posterior emission estimates and their uncertainties were governed by exploring the spaces of each of these parameters and hyper-parameters. The sensitivity of the emissions generally decreases with distance from the measurement site, which leads to increased uncertainty in the inversion, in both the spatial distribution of emissions and their overall magnitude. The further away emissions occur, the more likely the regional inversion method will allocate these emissions to a general diffuse region, rather than identify individual c-C4F8 point sources.
4.5 Regional model and inversion study using InTEM for western Europe
To investigate regional emissions in western Europe (36–66∘ N and −14–31∘ E) we used InTEM, an inversion framework (Arnold et al., 2018) based on the NAME Lagrangian transport model (Jones et al., 2007), together with observations from MHD, Tacolneston (TAC), Jungfraujoch (JFJ), and Monte Cimone (CMN). A priori estimates were considered unknown (see Sect. 3 and the Supplement) and therefore set to a uniform distribution of 0.2 Gg yr−1 over the whole land area within the inversion domain with an uncertainty of 0–0.62 Gg yr−1. Observational uncertainty was time varying and estimated as the variability of the observations in a 6 h moving window plus the measurement repeatability determined from repeat measurements of the on-site calibration standards. Model uncertainty was estimated every 2 h as the larger of the median of all pollution events at each station in a year or 16.5 % of the magnitude of the pollution event. A temporal correlation of 12 h was assumed in the model uncertainty at each station. An analytical solution was found that minimized the residual between the model and the observations and the difference between the posterior and the a priori flux estimate, balanced by the uncertainties of both. The baseline was estimated in the inversion following Arnold et al. (2018). The variable resolution of the inversion grid was calculated and refined within InTEM based on the magnitude of the footprint and emissions from each grid box. The inversions were run 24 times per year, each time with a randomly generated subsample (90 %) of the available observations from each station (10 % removed in 5 d blocks), to further explore the uncertainty. Emissions and uncertainties were averaged across the 24 individual inversions thereby assuming 100 % correlation between uncertainties in these separate inversions. We performed 1-year inversions covering the period 2013–2017.
4.6 Regional model and inversion study using NAME-HB for India
To investigate regional emissions from the Indian subcontinent from the samples taken on board a research aircraft in June and July 2016 (see Sect. 2.4) we used the NAME-HB inversion method described in Sect. 4.4 and Table S1. Here, the domain spanned from 6 to 48∘ N and from 55 to 109∘ E with an altitude up to 19 km and emissions were estimated as the mean over the 2-month period. As with eastern Asia and western Europe studies, the sensitivity of the atmospheric measurements to surface emissions was derived using the NAME model. Back-trajectories were simulated for each minute of each flight path for up to 30 d backward in time. To account for the motion of the aircraft, hypothetical air parcels were released from a cuboid whose dimensions were defined as the change in latitude, longitude, and altitude of the aircraft during each 1 min period, at a release rate of 1000 air parcels min−1. Wherever possible, samples were collected during periods of level flight, to minimize the altitude component of the release volume. India's a priori emissions were set to 18 % of global c-C4F8 emissions (from Sect. 5.2), equal to India's fraction of the global population, but uniformly distributed over India. A large uncertainty was assigned (Table S1 in the Supplement) to reflect the lack of information on India's current c-C4F8 emissions. A priori vertical boundary conditions were assigned using background mole fractions from MHD (N, E, and W) and CGO (S). Offsets to these boundary conditions were estimated in the inversion. Due to the limited number of samples taken on board the aircraft, the regional inversion for the Indian subcontinent may have more difficulty identifying individual point sources (see Sect. 4.4), which may also not be emitting at all times. We report only emissions for northern and central India (NCI) as the inversion has low sensitivity over southern India and Sri Lanka and the northwestern edge of the domain, and no sensitivity beyond the Himalayas (see Fig. S5). Sensitivity tests indicate that c-C4F8 emissions determined for NCI are insensitive to the choice of a priori emissions (see Fig. S6).
4.7 Pollution events at Zeppelin station
The Zeppelin (ZEP) station is located in a clean Arctic environment and receives air masses representative mostly of the Arctic background. Nevertheless, 10 cases of enhanced c-C4F8 mole fractions were observed with the arrival of air masses from Eurasia. To trace the origin of these events, we used 3-hourly 50 d backward simulations for a passive tracer with version 10 of the Lagrangian particle dispersion model FLEXPART (Stohl et al., 2005). The model was driven with operational meteorological analyses of the European Centre for Medium Range Weather Forecasts (ECMWF, https://www.ecmwf.int/, last access: 10 February 2019). The model set-up was similar to that typically used for inversion studies (Stohl et al., 2009), but the number of events observed at the station was too small for a sensible regional inversion. Instead, we inserted unit emission sources (∼1 kg s−1) at two facilities in Russia producing PTFE and halogenated chemicals including c-C4F8 (HaloPolymer, Kirovo-Chepetsk, Kirov Oblast and Galogen Open Joint-Stock Company, Perm), one or both of which we suspect to be responsible for the observed enhancements. We then scaled the modeled c-C4F8 mole fractions based on these two unit sources to the observed enhancements to estimate the source strength required to explain the observations. The two sources are quite close to each other and thus very much correlated so it was impossible to quantify the influence of each source individually, but it turned out that each source required about the same flux to produce a similar good match with the observations.
5.1 Atmospheric histories of c-C4F8 in both hemispheres
Figure 1 shows the atmospheric histories of c-C4F8 in the extratropical NH and SH determined from several sets of archive measurements and pollution filtered data from six in situ measurement stations. As detailed in Sect. 2.2, the data shown have gone through an iterative filtering process which mostly removed outliers from the NH record. The pollution-free monthly mean in situ data for the four extratropical NH stations shown here and ZEP agree within precisions, although JFJ data tend to be at the lower range since early 2015 for unknown reasons. The two extratropical SH stations CGO and ASA also agree well with each other. Mole fractions measured in both hemispheres show a clear and consistent interhemispheric gradient reflecting the high precision of the measurements and indicating that emissions of c-C4F8 predominantly occur in the NH. These data form a consistent atmospheric record of c-C4F8 from the late 1970s to 2017 in both hemispheres, albeit with very sparse data for the NH before in situ measurements started at JFJ and at other NH stations. The inset in Fig. 1 shows that the interhemispheric gradient, based on in situ measurements at high-latitude stations in the NH (MHD, THD, SIO) and SH (CGO) has been rising from ∼0.05 ppt in 2011 to ∼0.09 ppt in 2017, which suggests increasing, predominantly NH, emissions.
To augment our c-C4F8 data set and to extend our reconstruction further backwards in time, we measured air samples extracted at several firn sites from both hemispheres and interpreted the data with the CSIRO global inversion framework. The CSIRO inversion (see Sect. 4.3.1) yields the atmospheric history of c-C4F8 starting in 1900 until present, although abundances are essentially not different from zero (< 0.02 ppt) until the early 1960s (Fig. 3). Average global c-C4F8 mole fractions from the CSIRO inversion reached 0.45 ppt in 1980, 0.74 ppt in 1990, 0.97 ppt in 2000, 1.29 ppt in 2010, and 1.66 ppt in 2017. The Bristol inversion (see Sect. 4.3.2) does not incorporate firn data; still, atmospheric histories of the two inversions are generally in good agreement (see Fig. S7).
The CSIRO inversion reconstructs that the global rise rate of c-C4F8 accelerated from near zero before the late 1960s to ∼0.03–0.04 ppt yr−1 in the mid-1970s to late 1980s, after which the rise rate slowed to ∼0.02 ppt yr−1 in the early 1990s to mid-2000s. It increased again in the early 2000s and reached ∼0.07 ppt yr−1 in 2017.
Compared to Oram et al. (2012), our work extends the SH record from 2008 until present and, arguably, from 1978 back to 1900. Furthermore, it adds the full NH record. SH mole fractions reconstructed by Oram et al. (2012) are very similar in 1978 and 1990, but ∼0.06 ppt lower in the mid-1980s (∼11 %) and the late 1990s to late 2000s (∼5 %; see Fig. S8). Although the stated precision in Oram et al. (2012) of 0.8 % (∼0.01 ppt at 1.2 ppt) is similar to the 0.01–0.02 ppt achieved here, the resulting precisions of the CGAA measurements achieved here are significantly improved. For example, the noise in the CGAA reconstruction by Oram et al. (2012) is about as large as the interhemispheric gradient determined here (see Fig. S8). The estimated accuracy of the SIO-14 c-C4F8 calibration scale of ∼2 % also compares favorably to previous calibration scale uncertainties.
5.2 Global c-C4F8 emissions
Global c-C4F8 emissions (Fig. 5 and Supplement) started to increase in the early 1960s (CSIRO inversion) from near zero to ∼1.2 Gg yr−1 in the late 1970s to the late 1980s. The Bristol inversion initially reconstructs lower emissions, but the differences are within the estimated uncertainties for the reconstructed histories (see Fig. 5). Afterwards, emissions determined by both inversions declined to ∼0.8 Gg yr−1 in the mid-1990s to early 2000s. After that emissions kept increasing, reaching ∼2.2 Gg yr−1 in 2017. Both inversions reconstruct emissions which are significantly larger than available bottom-up inventory information (see Sect. 3 and the Supplement), reflecting the shortcomings of the current UNFCCC reporting requirements and bottom-up inventories.
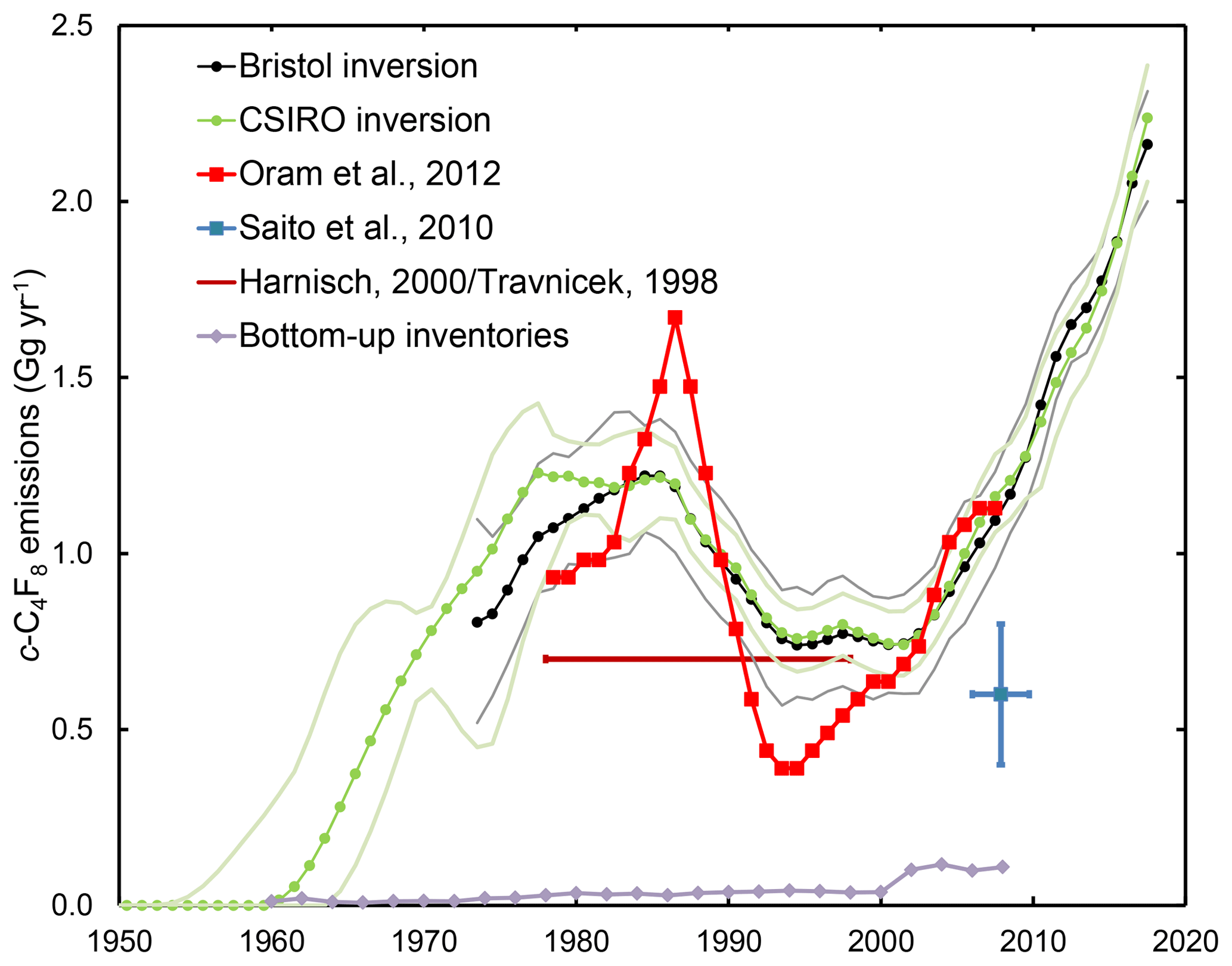
Figure 5Global c-C4F8 emissions reconstructed by the CSIRO inversion (green dots and line, light green 2σ uncertainty bands) from 1950 and by the Bristol inversion (black dots and line, grey 1σ uncertainty bands) from the early 1970s to present. In situ and archive data are used in both inversions, while firn air data are only used in the CSIRO inversion. Emission estimates by Oram et al. (2012) (red), Saito et al. (2010) (blue), Harnisch (2000)/Travnicek (1998) (brown), and from available bottom-up inventory information (grey) are shown for comparison.
Emissions presented by Oram et al. (2012) agree very well from 2001 to 2007 with our results and on average also from 1978 to 2001, although they show larger variability. Global emissions roughly estimated by Harnisch (2000) based on measurements by Travnicek (1998) of ∼0.7 Gg yr−1 from 1978 to 1997 are 30 % lower than our estimate of 1.01±0.10 Gg yr−1. Saito et al. (2010) estimated global emissions of 0.6±0.2 Gg yr−1 from January 2006 to September 2009, about half of our 1.16±0.09 Gg yr−1 estimate. This difference is likely due to slowly changing c-C4F8 mole fractions in calibration tanks used by NIES (Takuya Saito, personal communication, 2018), which would significantly affect the background rise rate and thus global emissions, but would have had less influence on the regional emissions estimated by Saito et al. (2010) as these are mostly dependent on the magnitude of the much larger pollution events above background.
Global emissions of c-C4F8 have clearly not leveled off at 2005–2008 levels as had been suggested by Oram et al. (2012), but kept rising. In contrast, emissions of other minor PFCs, C2F6 and C3F8, have decreased since the early 2000s and stabilized in recent years (Trudinger et al., 2016), reflecting that emission sources and/or use patterns of c-C4F8 are different from those of the other minor PFCs. Weighted by GWP100 (100-year timescale) estimated 2017 emissions of c-C4F8, C3F8, C2F6, and CF4 were 0.021, 0.005, 0.022, and 0.083 billion metric tons (tonnes) of CO2-eq., respectively (see Fig. S9). c-C4F8 CO2-eq. emissions have been larger than those of C3F8 since 2004 and, assuming continued growth, will also surpass C2F6 emissions within a year or two, so that c-C4F8 will become the second most important PFC emitted into the global atmosphere in terms of CO2-eq. emissions. In the next section, we will investigate regional emissions of c-C4F8 to gain a better understanding how individual regions and sources may contribute to the global emissions.
5.3 Regional c-C4F8 emission studies
5.3.1 Emissions from eastern Asia
Within the AGAGE network, the two stations in eastern Asia, Gosan (GSN) and Shangdianzi (SDZ), show by far the most frequent and most pronounced pollution events of up to ∼14 ppt above NH background, indicating significant regional emissions (see Fig. S10). Therefore, we use a regional inverse method (NAME-HB) to infer the emissions in this region (20–50∘ N and 110–160∘ E; see Sect. 4.4). We focus on the observations from GSN as this site was operated with relatively few interruptions from June 2010 to the end of 2017 and had almost full coverage for each year from 2011 to 2015. Significantly longer data gaps exist for SDZ, which would have made interpretation of inversion results more difficult. The sensitivity of the inversion generally decreases with distance to the receptor station, resulting in relatively low sensitivity for emissions from western China, eastern Japan, and parts of Taiwan (the cumulative footprint map for 2010–2017 is shown in Fig. S11). Therefore, we report in Table 2 and Fig. 6 estimated emissions for eastern China, western Japan, South Korea, North Korea, and Taiwan. c-C4F8 emissions in this eastern Asian domain increased from 0.36±0.07 Gg yr−1 in 2010 to 0.73±0.13 Gg yr−1 in 2016 and 2017 and were dominated by emissions from eastern China. The a priori emissions for eastern China of 0.185 Gg yr−1 are based on the Saito et al. (2010) estimate for all of China for November 2007 to September 2009, but the inversion suggests emissions that are ∼62 % higher in 2010 and more than triple in 2017. Note that if we were to sum up emissions for all regions of China, including those where the inversion has low sensitivity, total emissions would be another ∼50 %–75 % higher. In contrast, the EDGAR 4.2 emission inventory, the only available bottom-up information (see Sect. 3 and the Supplement), suggests no significant emissions from China.
Table 2Regional c-C4F8 emissions derived for eastern Asia from Gosan measurements (NAME-HB inversion) and comparison to global emissions (Gg yr−1, kt yr−1).
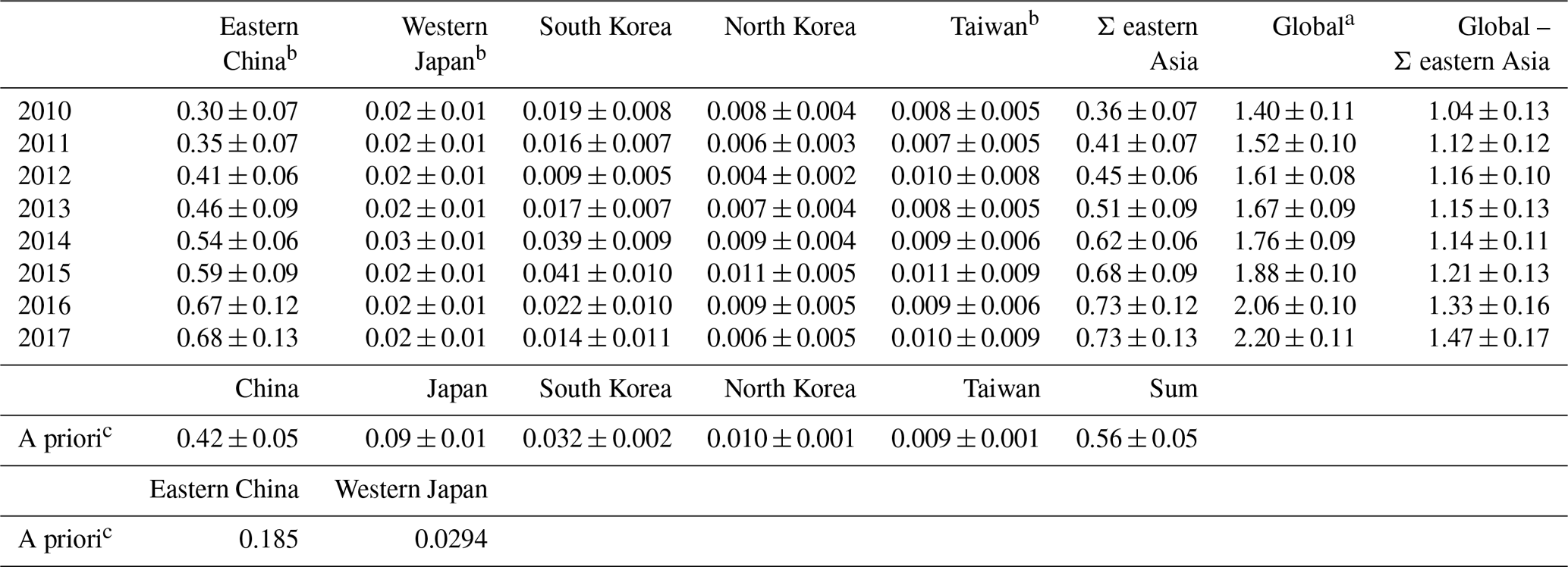
a Global emissions are the average of the emissions determined by the CSIRO and the Bristol inversion in this work. b Eastern China contains the provinces Anhui, Beijing, Hebei, Henan, Jiangsu, Liaoning, Shandong, Shanghai, Shanxi, Tianjin, and Zhejiang. Western Japan contains the prefectures Chugoku, Kansai, Shikoku, and Okawa and Kyushu. Due to the lower sensitivities of the inversion in western China, eastern Japan, and parts of Taiwan, where potential source industries are located, we cannot exclude further emissions in these regions and therefore total emissions are probably larger. c Saito et al. (2010) emission estimates based on atmospheric measurements from November 2007 to September 2009 were used as a priori information and were spread for each country uniformly over the area of each country. The resulting a priori estimates for eastern China and western Japan are additionally listed for comparison with the inversion results for these regions. Gosan measurements started in June 2010 with most complete coverages from 2011 to 2015.
For western Japan we find emissions of ∼0.02 Gg yr−1 (no trend), ∼30 % lower than the a priori emissions (from Saito et al. 2010; see Sect. 4.4). While total country emissions are likely higher, the available bottom-up information (see Sect. 3 and Supplement) suggests an order of magnitude lower emissions for all of Japan. For South Korea, the inversion adjusts emissions down to 0.01–0.02 Gg yr−1 in most years and up to ∼0.04 Gg yr−1 in 2014 and 2015. Except perhaps for 2012 and 2017, emissions from South Korea are significantly higher than the 0.003–0.008 Gg yr−1 suggested by the available bottom-up information. Emissions from Taiwan show no trend and are relatively small with ∼0.01 Gg yr−1, which is ∼50 % of ∼0.02 Gg yr−1 indicated by the Taiwanese NIR, though it should be noted that the inversion has relatively low sensitivities for some parts of Taiwan (see Fig. S11). Overall, emissions from western Japan, South Korea, and Taiwan are small, despite their large semiconductor industries (see also Fig. 7), suggesting that this industry sector is not a major emitter of c-C4F8. Emissions from North Korea are also small.
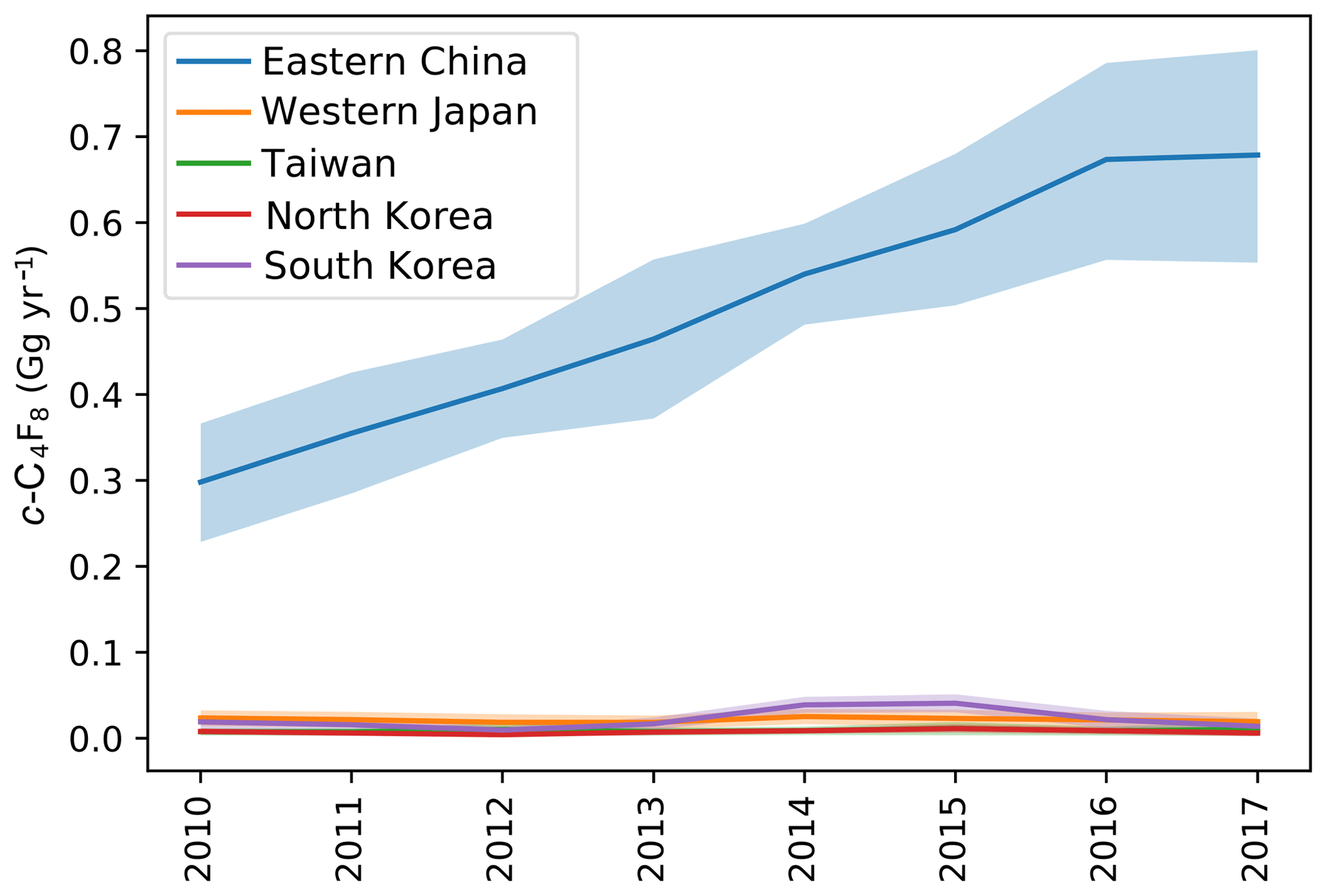
Figure 6c-C4F8 emissions in eastern Asia as determined by the NAME-HB regional inversion of measurements at the Gosan station, Jeju Island, South Korea, are dominated by emissions from China (blue). Emissions from western Japan (orange) and South Korea (violet) are much smaller; the latter show a small maximum in 2014 and 2015. Emissions from Taiwan (green) and North Korea (red) are also small. Shadings represent uncertainty bands of emissions.
Combined regional c-C4F8 emissions doubled from 2010 to 2016, driven by Chinese emissions. They represent 31±4 % of global emissions (2010–2017), while eastern China's emissions represent 28±4 %. The difference between global and eastern Asian emissions remained relatively consistent, ranging from ∼1.04 Gg yr−1 in 2010 to 1.47 Gg yr−1 in 2017 with an average of 1.20±0.14 Gg yr−1 from 2010 to 2017 and 1.15±0.03 Gg yr−1 from 2011 to 2015, the years with the best data coverage at GSN and thus highest confidence in the results. This means that the increase in global emissions is essentially explained by the increase in eastern Asian emissions, i.e., mostly from China, but also that significant emissions of ∼1.16 Gg yr−1 exist outside of the investigated region (a fraction of which may stem from industries located in parts of China and perhaps Japan where the inversion has low sensitivity).
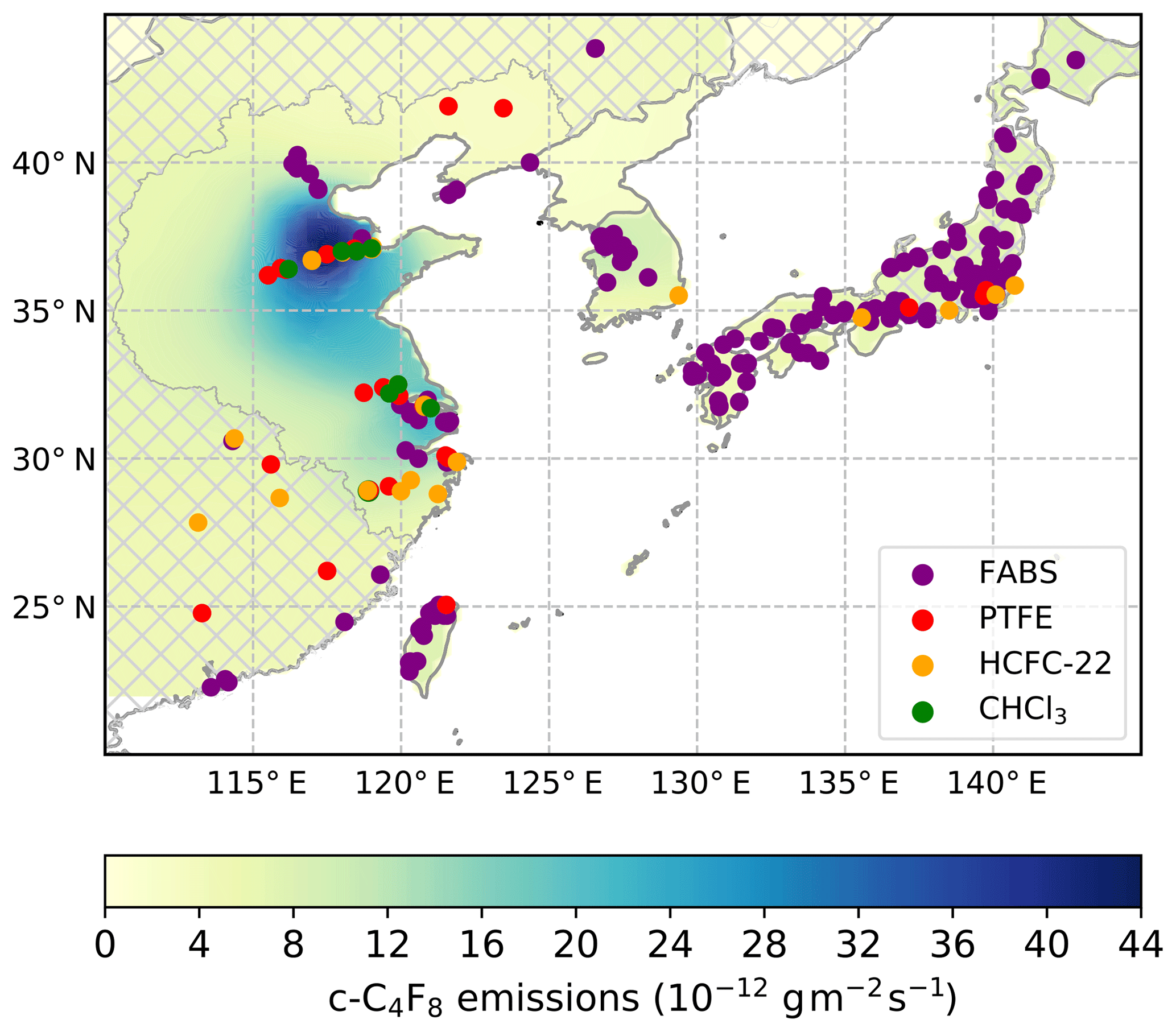
Figure 7Mean c-C4F8 emission strength (shades of green and blue, 10−12 g m−2 s−1) in eastern Asia from 2010 to 2017 determined by the NAME-HB inversion from measurements at the Gosan station, Jeju Island, South Korea. The hatching indicates areas for which emissions are not reported due to relatively low sensitivities of the inversion. Emissions predominantly occur in the densely industrialized Shandong, Tianjin, and parts of Henan and Hebei provinces south/southwest of Beijing as well as in Shanghai and neighboring provinces Jiangsu (to the north), Anhui (to the west), and Zhejiang (to the south) of the Yangtze River Delta region. Shown are industries with potential c-C4F8 emissions: semiconductor fabrication plants (FABS, purple dots, https://en.wikipedia.org/wiki/List_of_semiconductor_fabrication_plants, last access: 3 December 2018; http://www.10stripe.com/featured/map/semiconductor-fabs.php, last access: 3 December 2018; and other sources) and TFE–HFP–PTFE–FEP production facilities (PTFE, red dots, https://www.qianzhan.com/analyst/detail/220/170629-c33a2ca7.html, last access: 12 December 2018; and other sources). HCFC-22 (orange dots) and chloroform (CHCl3, green dots) production facilities are shown as the TFE and HFP monomers needed to produce PTFE and FEP fluoropolymers are produced via pyrolysis of HCFC-22 and c-C4F8 is an intermediate/by-product in this process, while HCFC-22 is manufactured from CHCl3.
Figure 7 shows that from 2010 to 2017 emissions in eastern China occur from the highly industrialized provinces Shandong, Tianjin, and parts of Henan and Hebei (south/southwest of Beijing) as well as from Shanghai and neighboring Jiangsu (to the north), Anhui (to the west), and Zhejiang (to the south) in the Yangtze River Delta region. Also shown are locations of potential industrial c-C4F8 point sources. For South Korea, western Japan and Taiwan, semiconductor fabrication plants do not seem to be dominant c-C4F8 emitters as they are not co-located with large c-C4F8 emissions (though the inversion has low sensitivity for eastern Japan, where many more semiconductor fabrication plants (FABS) and several PTFE and HCFC-22 plants are located; hence emissions from this region cannot be analyzed).
In China, the picture is less clear than in South Korea, Japan, and Taiwan, as several semiconductor fabrication plants in the Yangtze River Delta region are co-located with strong c-C4F8 emissions, while those near Beijing are not. Many of the potential production facilities of TFE and HFP monomers and PTFE and FEP polymers are co-located with areas where strong c-C4F8 emissions occur. This is consistent with information from the second largest producer of PTFE in China that they do not recover c-C4F8 by-product, but do emit c-C4F8 to the atmosphere (Jianxin Hu, personal communication, 2018). Still, the two facilities northeast of Beijing do not seem to emit c-C4F8, perhaps reflecting that some producers minimize c-C4F8 emissions, e.g., to increase yield or to use c-C4F8 for other purposes, such as for the semiconductor industry. Several facilities are also located in provinces for which the inversion has low sensitivity. Most HCFC-22 production facilities are not co-located with strong c-C4F8 emissions, while CHCl3 production facilities tend to be in areas with c-C4F8 emissions. This may reflect that CHCl3 production has shifted from use as a feedstock to produce HCFC-22 for dispersive applications (refrigeration or foam blowing), where no c-C4F8 emissions occur, to production of TFE–HFP–PTFE–FEP via HCFC-22 pyrolysis, where c-C4F8 by-product emissions occur, perhaps at the same or close-by facilities. This would be consistent with the start of the HCFC phase-out for dispersive applications in developing countries mandated by the Montreal Protocol on Substances that Deplete the Ozone Layer. Then again, CHCl3 has other uses, e.g., as solvent (Tsai, 2017), without any potential c-C4F8 emissions.
Note that at one or both of the PTFE production facilities in Zhejiang province (Juhua Group Corporation) HFO-1234yf has been produced since 2016, using a process which starts out with the same chemistry needed for PTFE–FEP production, that is the pyrolysis of HCFC-22 to TFE and HFP, with c-C4F8 as a potential by-product (see Supplement).
There is no strong correlation between c-C4F8 emission distribution and population density, e.g., emissions from Henan and Hebei provinces are significantly lower than those from Shandong despite similar total population, which may indicate that combustion of fluoropolymers in waste incineration facilities (Morisaki, 1978; Kannan et al., 2005; van der Walt et al., 2008; Ji et al., 2016; Bezuidenhoudt et al., 2017) is not a dominant source of c-C4F8 emissions.
If c-C4F8 emissions in eastern Asia are predominantly associated with TFE–HFP–PTFE–FEP production via the pyrolysis of HCFC-22, c-C4F8 emissions may co-occur with small emissions of TFE and HFP. HFC-23 emissions may also co-occur as HCFC-22 is produced from CHCl3 and HFC-23 is a by-product that in developing countries is probably again vented to the atmosphere since the UNFCCC Clean Development Mechanism (CDM) funding to avoid HFC-23 emissions has expired (Simmonds et al., 2018; Say et al., 2019). While the global atmospheric lifetime of TFE is only ∼2 d, the lifetime of HFP is ∼6 d (Acerboni et al., 2001), so that HFP may be detectable near strong emission sources and serve as a sensitive marker for regional TFE–HFP–PTFE–FEP production. After adding HFP to the measurements in late 2018, we find that HFP pollution events at SDZ always coincide with c-C4F8 and HFC-23 pollution events (see Fig. S12 and its caption in the Supplement). HFP pollution events at GSN are much weaker, reflecting the short atmospheric lifetime and the more distant source region, but they also coincide with c-C4F8 and HFC-23 pollution events. At both sites, however, c-C4F8 pollution events also coincide with enhancements of other anthropogenic compounds which may just point to generally polluted air in the region, so it is difficult to draw definitive conclusions. Still it is clear that HFP is emitted in eastern Asia, likely in China, and HFP as well as c-C4F8 emissions can be explained by PTFE–FEP production. Measurements of HFP at SIO and ASA confirm that it is virtually absent (≤0.01 ppt) from the global background atmosphere even in urban environments.
Overall, the strong c-C4F8 emissions in eastern China and their source regions are consistent with our hypothesis of emissions from TFE–HFP–PTFE–FEP production facilities due to little or no recovery or abatement of c-C4F8 by-product and the significant fraction of global PTFE production (53 %–67 % in 2015) occurring in China (see Table S3).
5.3.2 Emissions from northwestern Europe
Outside of eastern Asia, the TAC station in East Anglia, UK, shows by far the most frequent and most pronounced c-C4F8 pollution events of any AGAGE station, with a few reaching ∼5 to 10 ppt above NH background, indicating close-by emissions. Data from the TAC, MHD, JFJ, and CMN stations and the InTEM regional inverse method (see Sect. 4.5) were used to estimate emissions from northwestern Europe (42 to 59∘ N and −11 to 15∘ E) based on the areas of highest sensitivity to the observations (see Fig. S13). Compared to eastern Asia, we find only small emissions of 0.026±0.013 Gg yr−1 (Ireland, UK, France, Germany, Belgium, the Netherlands, Luxemburg, and Denmark, 2013–2017) without any significant temporal trend, corresponding to only ∼1 % of global emissions, despite an estimated 14 % of global PTFE production in 2015 (see Table S3). The mean distribution of emissions is shown in Fig. 8. Similar to eastern Asia, many identified semiconductor FABS in Europe are not co-located with c-C4F8 emission hot spots, while several FABS in northern France, the UK, Ireland, and the Netherlands seem to be co-located. Producers of PTFE and FEP and facility locations in Europe were determined from company websites (3M/Dyneon, AGC/Asahi Glass, Arkema, Chemours/DuPont, Saint-Gobain, Solvay) and the European Pollutant Release and Transfer Register (https://prtr.eea.europa.eu/#/home, last access: 16 January 2019), but it is very difficult to determine at which of the many facilities PTFE or FEP is actually produced and thus where c-C4F8 may be emitted. It seems that several facilities in the Netherlands, Belgium, the UK, France, and Italy which likely produce PTFE are co-located with identified c-C4F8 emission hot spots (Fig. 8). Still, many mismatches exist, reflecting the uncertainties in determining the exact facility locations, the relatively small emission strength, and uncertainties of the inversion. As in eastern Asia, there seems to be no correlation with population density, which suggests that waste incineration of fluoropolymers is not a dominant c-C4F8 source here either. The inversion is broadly consistent with emissions from TFE–HFP–PTFE–FEP production and FABS, but emissions from other industrial sources may also play a role. While emissions of 0.026±0.013 Gg yr−1 are relatively small, it is noteworthy that UNFCCC reports suggest much smaller c-C4F8 emissions (0.0007 Gg yr−1, UNFCCC, 2013–2014 and 0.0017 Gg yr−1, bottom-up emission inventories, Sect. 3, 2013–2014).
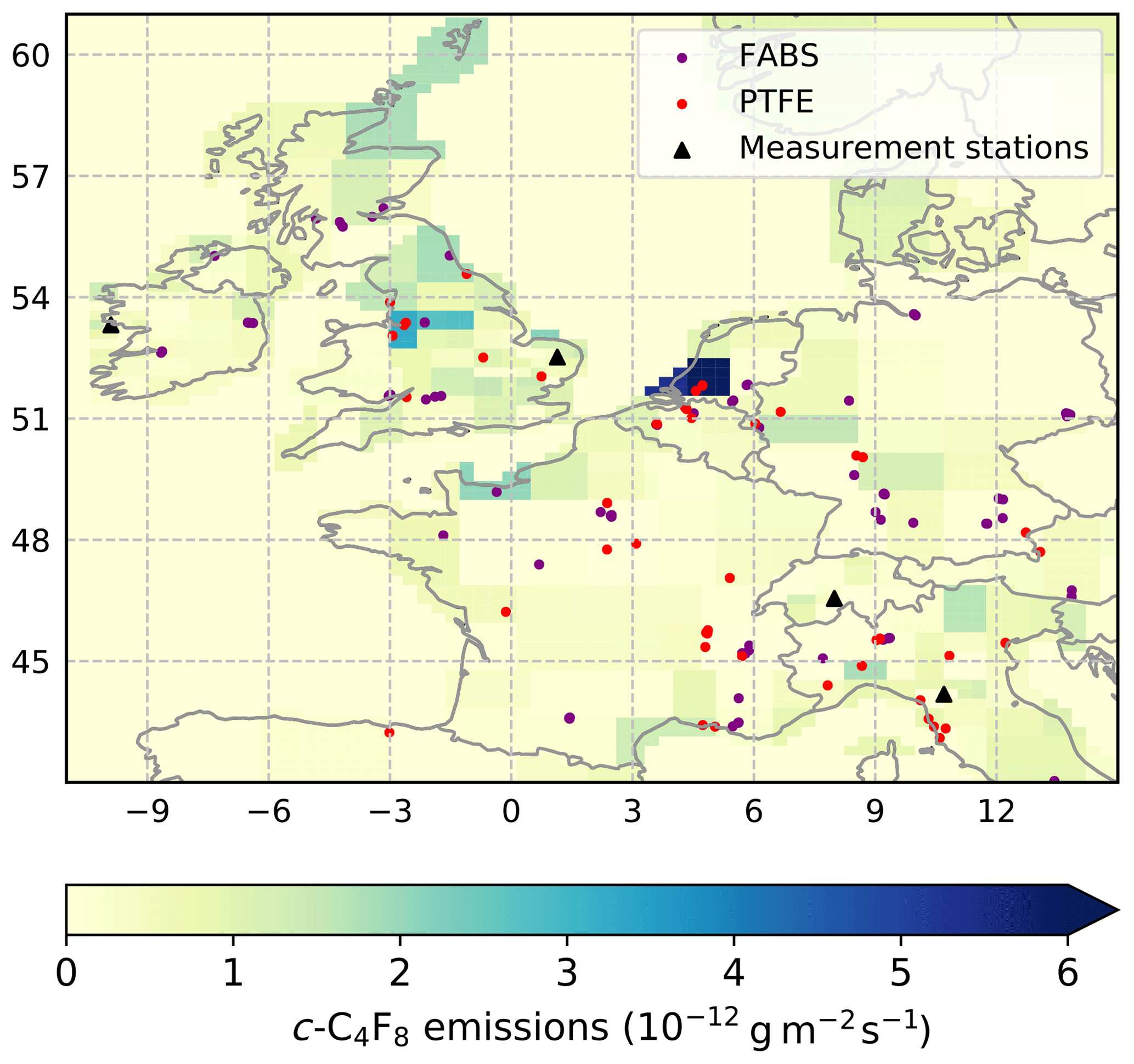
Figure 8Mean c-C4F8 emission strength (shades of green and blue, 10−12 g m−2 s−1) in northwestern Europe (42 to 59∘ N and −11 to 15∘ E) from 2013 to 2017 determined by the InTEM inversion from measurements at four sites (Mace Head, Ireland, Tacolneston, United Kingdom, Jungfraujoch, Switzerland, and Monte Cimone, Italy, black triangles). Also shown are potential industrial emitters of c-C4F8. Locations of potential TFE/HFP/PTFE/FEP production facilities (red dots) are based on company websites (3M, Chemours, Daikin, DuPont, Saint-Gobain, and Solvay) and are much less certain than the corresponding location information for eastern Asia. Also shown are semiconductor fabrication plants (purple dots, https://en.wikipedia.org/wiki/List_of_semiconductor_fabrication_plants, last access: 3 December 2018; http://www.10stripe.com/featured/map/semiconductor-fabs.php, last access: 3 December 2018; and other sources).
5.3.3 Emissions from southeastern Australia
Other urban locations of the AGAGE network, such as SIO, USA, and ASA, Australia, show much smaller pollution events above global background (up to ∼2.5 ppt) than those seen at TAC, suggesting even lower emissions. Still, the few pollution events at ASA and even CGO are interesting as production of PFCs in Australia has never been recorded. CFC-11, CFC-12, and HCFC-22 were manufactured starting in 1962 at two facilities in Sydney, but production ceased in 1995 and trace gas emissions from Sydney are rarely if ever observable at CGO or ASA. Without any currently known fluorocarbon production, any c-C4F8 pollution events observed at CGO or ASA should not be due to fugitive emissions. c-C4F8 imports to Australia are ∼4 to 50 kg yr−1 (2011–2015), likely for minor refrigeration uses. In contrast, small but identifiable c-C4F8 pollution episodes at CGO suggest Melbourne emissions of ∼2 t yr−1 (0.002 Gg yr−1) in 2016 (down from ∼5 t yr−1 in 2009, inter-species correlation method, ISC; see Fraser et al., 2014; Dunse et al., 2018). Scaled by population to Australia (for lack of a better proxy), emissions from 2009 to 2016 could be ∼10–25 t yr−1 (0.01–0.025 Gg yr−1), 2–3 orders of magnitude higher than import data suggest. Since early 2017, HFP has been measured at ASA (see Fig. S12 caption). Occasionally, small HFP pollution events, which are often, but not always, associated with c-C4F8 pollution events, may point to small-scale production of TFE–HFP–PTFE–FEP in Melbourne or perhaps these small emissions stem from incineration of waste-containing fluoropolymers. Another possible explanation could be that more c-C4F8 is imported in products for minor applications than identified in import data due to inadequate labeling. On a global scale, estimated Australian c-C4F8 emissions of ∼0.015 Gg yr−1 are small, ∼0.7 % of global emissions. PFC (CF4, C2F6) pollution episodes at Cape Grim and Aspendale due to PFC emissions from southeastern Australian aluminum smelters (Portland and Pt. Henry, Victoria, and Bell Bay, Tasmania) do not show any evidence of c-C4F8 emissions (Fraser et al., 2013; CSIRO unpublished data).
5.3.4 Emissions from under-sampled regions such as the US, India, and Russia
The AGAGE network does not closely monitor large areas of the globe where c-C4F8 emissions may occur. For example, many semiconductor FABS are located in the western, southern, and eastern US and chemical facilities located in the southern and eastern US are estimated to account for ∼10 % of global PTFE production in 2015, while facilities in India and Russia are estimated to account for ∼8 % and ∼6 %, respectively (see Tables S3 and S4). The two AGAGE stations in California are only able to capture a fraction of these emissions due to predominant westerly winds and therefore we cannot estimate c-C4F8 emissions from the continental US. If PTFE production facilities in the US are operated as in NW Europe, emissions should be similarly small. If facilities in India and Russia are operated as in China, emissions could be significant as well. In the case of Russia this seems likely as the original technology for fluoropolymer production in China apparently stems from Russia (Buznik, 2009).
5.3.5 Emissions from India
Say et al. (2019) recently presented measurements from an aircraft campaign in June and July 2016 (see Sect. 2.4) over the Indian subcontinent to determine emissions of ODS and HFCs. Here we use their c-C4F8 measurements and the NAME-HB inversion (see Sect. 4.6) and estimate emissions of 0.14 (0.09–0.20) Gg yr−1 for northern and central India (NCI). Data are only available for two months in 2016, but seasonality in industrial emissions of c-C4F8 is not expected. Given the limitations of the inversion method to identify distant point sources from a relatively small number of samples (see Sect. 4.4 and 4.6), the posterior emission distribution (Fig. 9) is consistent with emissions from facilities producing PTFE. Several of the HCFC-22 production facilities are co-located or very close to these PTFE-producing facilities, suggesting that a fraction of HCFC-22 is pyrolyzed to produce monomers for PTFE and FEP. Two HCFC-22 production facilities are outside of areas with strong c-C4F8 emissions, possibly because these two sites focus on production of HCFC-22 for dispersive applications (refrigeration or foam blowing), where no c-C4F8 emissions occur. The single FAB in India we are aware of is not co-located with significant c-C4F8 emissions. As in eastern Asia and northwestern Europe, there is no apparent correlation of c-C4F8 emissions with population density. Emissions predominantly occur outside of the Indo-Gangetic plain, the most densely populated region of India, which excludes potential sources that scale with population. Instead the inversion allocates emissions in a much less densely populated region in which multiple likely industrial point sources for c-C4F8 are located. The derived emissions account for 6.8 (4.4–9.7) % of global c-C4F8 emissions in 2016, in comparison to the estimated ∼8 % of 2015 global PTFE production capacity (see Table S3). While we cannot categorically exclude an unknown industrial source, these results are consistent with the chemistry of TFE–HFP–PTFE–FEP production as a dominant c-C4F8 emission source. Note that one of the facilities in western India (Navin Fluorine International, Surat, Gujarat) is known to also produce HFO-1234yf since 2016, using a process which starts out with the same chemistry, that is the pyrolysis of HCFC-22 to TFE and HFP, with c-C4F8 as potential by-product (see Supplement). All known Indian PTFE manufacturers are located within the NCI domain; hence the estimated emissions are likely to be roughly representative of India's national total, though further atmospheric measurements would be required to confirm this.

Figure 9Mean c-C4F8 emission strength (shades of green and blue, 10−14 g m−2 s−1) over the Indian subcontinent for June and July 2016 determined by the NAME-HB inversion based on air samples taken on board UK's FAAM (Facility for Airborne Atmospheric Measurements) BAe-146 research aircraft. Also shown are the locations of one semiconductor fabrication plant (FAB, purple dot) and several potential TFE–HFP–PTFE–FEP production facilities (PTFE, red dots, Solvay/CYTEC, Hindustan Fluorocarbons, and Gujarat Fluorochemicals facilities) as potential c-C4F8 sources. HCFC-22 (orange dots) production facilities are also shown as the TFE and HFP monomers needed to produce PTFE and FEP fluoropolymers are produced via pyrolysis of HCFC-22 and c-C4F8 is an intermediate/by-product in this process. The outline of the northern and central India (NCI) model domain is shown as a pink line.
5.3.6 Emissions from facilities in Russia
Measurements at the ZEP site in remote Svalbard show 10 small c-C4F8 pollution events above the NH background of up to ∼0.4 ppt. FLEXPART backward simulations could trace some of these events to two facilities in Russia which produce PTFE and halogenated chemicals including c-C4F8 itself (HaloPolymer, Kirovo-Chepetsk, Kirov Oblast, and Galogen Open Joint-Stock Company, Perm). Figure S14 shows the FLEXPART footprint emission sensitivity map for the largest observed c-C4F8 enhancement on 19 November 2016, suggesting direct transport from the two sites. The emission sensitivity maps indicate that for six of the 10 observed pollution events the air had clearly passed over one or both of these two sources, even though the timing of the observed events was often not well matched by the model, which was sometimes off by up to about half a day. While this is not surprising given the large distance between the source and the receptor, it means that the two sources could not be clearly separated, especially since the FLEXPART emission sensitivity often also covered both sites for the same arrival times at ZEP. Assuming a unit emission at those two locations and scaling the resulting simulated mole fractions at ZEP to the observed enhancements above background, we estimated the emission strength for the two sites together for each event (see Sect. 4.7). Five of the 10 pollution events could be approximately reproduced by this method and required a flux of 0.18±0.06 Gg yr−1, while the sixth event required ∼0.54 Gg yr−1. Averaged for all six events 0.24±0.15 Gg yr−1 would be required. Either of these fluxes would be significant, representing 9±3 %, 26 %, and 12±7 % of global emissions, respectively, compared to ∼6 % of estimated global PTFE production in Russia. The uncertainty of this estimate is large because only a few events were observed and not all of them were reproduced equally well by FLEXPART. Similar to eastern Asia, the largest c-C4F8 pollution event also showed enhancements of HFC-23, pointing to TFE–HFP–PTFE chemistry as a source (see Fig. S15), but other halogenated compounds were also elevated.
We determine the atmospheric histories of c-C4F8 (PFC-318, perfluorocyclobutane) in both hemispheres based on measurements of archived, in situ, and firn air samples in conjunction with the CSIRO firn model, the AGAGE 12-box model, and two global inversion frameworks. Compared to previous studies, our work extends the Southern Hemisphere record from 1978 back to 1900 and from 2008 until 2017 and adds a Northern Hemisphere record, all reported with improved precisions for air archive measurements (∼1 %–2 %) and a lower uncertainty (2 %) of the SIO-14 gravimetric calibration scale. We find global c-C4F8 atmospheric mole fractions near zero (< 0.02 ppt) from 1900 until the early 1960s, after which they rose sharply, reaching 0.45 ppt in 1980, 0.74 ppt in 1990, 0.97 ppt in 2000, 1.29 ppt in 2010, and 1.66 ppt in 2017. Global c-C4F8 emissions started to increase in the 1960s from near zero to (1σ) Gg yr−1 in the late 1970s to the late 1980s. After this, emissions declined to Gg yr−1 in the mid-1990s to early 2000s. After this emissions again increased, reaching ∼ 2.20±0.05 Gg yr−1 in 2017. These global emissions are significantly larger than what can be compiled from available bottom-up inventory information (70±17 times, 1990–1996, 29±5 times, 1997–2010, 15±1 times, 2011–2014).
Using the NAME-HB regional inverse method and observations at Gosan station we find that emissions from eastern Asia rose from ∼0.36 Gg yr−1 in 2010 to ∼0.73 Gg yr−1 in 2016 and 2017, representing 31±4 % of global emissions, predominantly from eastern China. Strong c-C4F8 emissions are found from heavily industrialized provinces south/southwest of Beijing and near the Yangtze River Delta. In contrast, emissions from western Japan, South Korea, and Taiwan are small, suggesting that their large semiconductor industries are not major c-C4F8 emitters. While we cannot categorically exclude emissions from other industrial sources, overall, the strong c-C4F8 emissions in eastern China and their spatial pattern are roughly consistent with emissions from production of TFE–HFP–PTFE–FEP and other fluorochemicals. A significant fraction of global PTFE production (53 %–67 % in 2015) occurs in China and, as indicated by the second largest producer of PTFE in China, the c-C4F8 by-product from the underlying HCFC-22 pyrolysis process is not recovered or abated, but rather emitted to the atmosphere.
Based on samples collected over the Indian subcontinent in mid-2016, we determine emissions of 0.14 (0.09–0.20) Gg yr−1 c-C4F8 from northern and central India (NCI), ∼6.8 (4.4–9.7) % of global emissions. Within the limitations of the inversion, the determined emission map is also roughly consistent with emissions from TFE–HFP–PTFE–FEP production.
Using the InTEM regional inverse method and measurements at four western European stations, we only find small NW European emissions of Gg yr−1 c-C4F8 from 2013 to 2017 (∼1 % of global emissions), in contrast to an estimate of 14 % of global PTFE production capacity in 2015. The inversion also points to facilities which may produce PTFE and FEP and semiconductor fabrication plants though the picture is less clear.
No obvious correlation between population density and c-C4F8 emissions is found in eastern Asia, NCI, and NW Europe, indicating that incineration of waste-containing fluoropolymers is not a major source of c-C4F8.
Based on data from two Australian stations and an inter-species correlation method, Australian c-C4F8 emissions are estimated to be small, perhaps ∼0.7 % of global c-C4F8 emissions. We find no evidence for c-C4F8 production from three large aluminum smelters in SE Australia.
Based on a few c-C4F8 pollution events observed at Zeppelin station and a rough FLEXPART analysis, we estimate that emissions from two Russian facilities known to produce PTFE and halocarbons including c-C4F8 itself could be Gg yr−1. While this could represent a significant fraction of global emissions (possibly ranging from 5 % to 26 %), uncertainties are very large.
In summary, for the year 2016, we find global c-C4F8 emissions of 2.06±0.10 Gg yr−1, with 0.73±0.12 Gg yr−1 from parts of eastern Asia (36 % of the global total), 0.14 (0.09–0.20) Gg yr−1 from northern and central India (6.8 %), Gg yr−1 from northwestern Europe (∼1 %), and ∼0.015 Gg yr−1 from Australia (∼ 0.7 %).
Current monitoring capabilities of the AGAGE network leave large areas with potential c-C4F8 emission sources un- or under-monitored, e.g., most of the US, India, Russia, western China, and eastern Japan, where various semiconductor facilities and fluorochemical and fluoropolymer production plants are located.
While many possible uses and emission sources of c-C4F8 are found in the literature and though we cannot categorically exclude unknown industrial sources, the start of significant c-C4F8 emissions around the 1960s may well be related to the initial synthesis of PTFE in 1938 with commercial production of PTFE (Teflon) by DuPont commencing in 1947 (Gangal and Brothers, 2015) via pyrolysis of HCFC-22, with c-C4F8 as a by-product/intermediate. It seems unlikely that process control or abatement to minimize c-C4F8 by-production was in place in the early decades of PTFE production and c-C4F8 by-product was probably emitted to the atmosphere, explaining the steep increase in global emissions reconstructed here. With the advent of UNFCCC by-product reporting requirements in the 1990s, concern about climate change and product stewardship, abatement, and perhaps collection of c-C4F8 for use in the semiconductor industry where it can be easily abated, it is conceivable that fugitive c-C4F8 emissions in developed countries (UNFCCC Annex 1) overall were reduced, explaining the observed stabilization and reduction of global emissions in the 1980s and 1990s. Similar efforts to contain and destroy by-product emissions of fluorocarbons, e.g., HFCs, from the 1980s to the 2000s are documented in the Toxics Release Inventory (https://www.epa.gov/toxics-release-inventory-tri-program, last access: 5 February 2019) Program of the U.S. EPA and the European Pollutant Release and Transfer Register. Concurrently, production of PTFE in China increased rapidly, e.g., from 2000 to 2005 by ∼ 26 % yr−1, followed by a slowdown to ∼14 % yr−1 from 2005 to 2015 and perhaps ∼8 % yr−1 from 2015 onward, reaching an estimated 53 %–67 % of global production in 2015 (see Tables S2, 3, and 4). Without any emission reduction requirements, it is conceivable that fugitive emissions of c-C4F8 from TFE–HFP–PTFE–FEP production in China, and other developing (UNFCC non-Annex 1) countries today dominate global emissions. The 2010 to 2016 rise in rates of eastern Chinese (eastern Asian) c-C4F8 emissions of ∼15 % yr−1 (∼13 % yr−1) determined here is compatible with these PTFE production increase rates of 14 to 8 % yr−1 in China. Barring other developments, we predict that c-C4F8 emissions will continue to rise and that c-C4F8 will become the second most important PFC emitted to the global atmosphere in terms of CO2-eq. emissions within a year or two. While the 2017 radiative forcing of c-C4F8 (∼0.52 mW m−2) is very small compared to that of CO2, emissions of c-C4F8 and other perfluorinated compounds with similarly long lifetimes and high radiative efficiencies essentially permanently alter the radiative budget of Earth. The fact that significant emissions of ∼1.16 Gg yr−1 of global emissions (56 %) exist outside of the monitored regions clearly shows that observational capabilities and reporting requirements need to be improved to understand global and country-wide emissions of PFCs and other synthetic greenhouse gases and ozone-depleting substances.
AGAGE in situ data are available at http://agage.mit.edu/data (last access: 10 February 2019), http://cdiac.ess-dive.lbl.gov/ndps/alegage.html (last access: 10 February 2019), and https://data.ess-dive.lbl.gov/view/doi:10.3334/CDIAC/ATG.DB1001 (last access: 10 February 2019). For CSIRO and Bristol inversion results, firn data, etc. see the Supplement. Please contact Jens Mühle (jmuhle@ucsd.edu) for further information.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-10335-2019-supplement.
JM contributed to archive, firn, and in situ measurements, interpreted the data, and prepared the paper with contributions from all co-authors. CMT provided CSIRO firn model and CSIRO global inversion results and interpretation. MR provided Bristol global inversion results. LMW provided NAME-HB model runs and emissions estimated for East Asia; DS and ALG provided the same for India. AJM and LMW provided InTEM model runs and emissions for Europe. AS and NE provided FLEXPART model runs and guided estimation of Russian emissions. DS and ALG provided the aircraft data from India. CMT, MR, LMW, AJM, DS, ALG, AS, and PJF contributed significantly to the text. LPS, DJI, TA, JM, PJF, and PBK provided and oversaw CSIRO air archive and NH archive measurements. MKV, SP, SL, MKP, COJ, LPS, PBK, SOD, PGS, DY, PBK, KMS, OH, BM, CL, JK, JA, MM, SR, and BY oversaw station operations and provided quality-controlled measurement data. PJF provided the estimate of Australian emissions. CMH provided gravimetric calibration and calibration propagation for the whole AGAGE network. PKS wrote the software to run all instruments and analyze all measurement data. MKV, BH, CB, VP, DME, and JS provided firn data and were instrumental in their interpretation. AMC provided insight into UNFCCC reporting and bottom-up inventories as well as industrial processes. EM and MC greatly helped with the gathering of locations of semiconductor facilities. RGP and RFW provided overall project oversight.
The authors declare that they have no conflict of interest.
Development of the Medusa GC–MS systems, calibrations, and measurements at the Scripps Institution of Oceanography, La Jolla, as well as operations of Trinidad Head, CA, were carried out as part of the international AGAGE research program and supported by the NASA Upper Atmospheric Research Program in the US with grants NNX07AE89G and NNX16AC98G to MIT and NNX07AF09G, NNX07AE87G, NNX16AC96G, and NNX16AC97G to SIO. In the UK, the Department for Business, Energy & Industrial Strategy (BEIS) provided support through contract 1028/06/2015 to the University of Bristol for Mace Head, Ireland, for Tacolneston, UK, and to the UK Met Office for InTEM analysis. The National Oceanic and Atmospheric Administration (NOAA) in the US provided support to the University of Bristol for operations at Ragged Point, Barbados, through contract RA-133-R15-CN-0008 and supported operations at Cape Matatula, American Samoa. Operations in Australia were supported by the Commonwealth Scientific and Industrial Research Organization (CSIRO), the Bureau of Meteorology (Australia), the Department of Environment and Energy (Australia), and Refrigerant Reclaim Australia. Operations at Jungfraujoch were supported by the Swiss National Program HALCLIM/CLIMGAS-CH (Swiss Federal Office for the Environment, FOEN) and by the International Foundation High Altitude Research Stations Jungfraujoch and Gornergrat (HFSJG). Operations at Zeppelin were supported by the Norwegian Environment Agency. Operations at Monte Cimone were supported by the National Research Council of Italy and the Italian Ministry of Education, University and Research through the Project of National Interest NextData. Operations at Gosan were supported by the National Strategic Project-Fine particle of the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (MSIT), the Ministry of Environment (ME), and the Ministry of Health and Welfare(MOHW) (no. NRF-2017M3D8A1092225). Operations at Shangdianzi were supported by the National Nature Science Foundation of China (41575114). We are indebted to the staff and scientists at AGAGE and other sites for their continuing contributions to produce high-quality measurements of atmospheric trace gases. Firn air sampling at Law Dome was supported by the Australian Antarctic Division, Australian Antarctic Science Program, and Australia's Nuclear Science and Technology Organisation. We acknowledge the members of the firn air sampling teams at South Pole in 2001 and at NEEM in 2008. Firn air sampling at the Summit station was supported through NSF grants ARC-1203779 and ARC-1204084, with airlift support from the 109th Airlift Wing of the New York Air National Guard. We thank Ed J. Dlugokencky and the National Oceanic and Atmospheric Administration (NOAA) Earth System Research Laboratory (ESRL) Global Monitoring Division (GMD) Carbon Cycle Greenhouse Gases (CCGG) group for measurements which were instrumental for characterizing the Summit13 firn site. We also thank Dave Keeling (deceased) and Ralph Keeling (SIO) for air samples. We thank Takuya Saito for helpful discussions. Matthew Rigby was supported in part by advanced research fellowships from the UK Natural Environment Research Council (NERC, NE/1021365/1). Luke Western was supported by NERC grant NE/M014851/1. Anita L. Ganesan was funded under a NERC Independent Research Fellowship (NE/L010992/1). We acknowledge the contribution of NERC, the Ministry of Earth Sciences, Government of India, and the principal investigators of “Drivers of the South Asian Monsoon” aircraft campaign in India. Funding for the measurements used here was made possible by NERC grant NE/I027282/1. We thank the two anonymous reviewers for their very helpful comments.
This research has been supported by the National Aeronautics and Space Administration (grant nos. NNX07AE89G, NNX07AF09G, and NNX07AE87G), the UK Department for Business, Energy & Industrial Strategy (BEIS) (grant no. 1028/06/2015), the National Oceanic and Atmospheric Administration (grant no. RA-133-R15-CN-0008), the National Natural Science Foundation of China (grant no. 41575114), the National Science Foundation (grant nos. ARC-1203779 and ARC-1204084), and the Natural Environment Research Council (grant no. NE/I027282/1). Further information is given in the Acknowledgements.
This paper was edited by Farahnaz Khosrawi and reviewed by two anonymous referees.
Acerboni, G., Beukes, J. A., Jensen, N. R., Hjorth, J., Myhre, G., Nielsen, C. J., and Sundet, J. K.: Atmospheric degradation and global warming potentials of three perfluoroalkenes, Atmos. Environ., 35, 4113–4123, https://doi.org/10.1016/S1352-2310(01)00209-6, 2001.
Adolph, A. C. and Albert, M. R.: Gas diffusivity and permeability through the firn column at Summit, Greenland: measurements and comparison to microstructural properties, The Cryosphere, 8, 319–328, https://doi.org/10.5194/tc-8-319-2014, 2014.
Arnold, T., Mühle, J., Salameh, P. K., Harth, C. M., Ivy, D. J., and Weiss, R. F.: Automated measurement of nitrogen trifluoride in ambient air, Anal. Chem., 84, 4798–4804, https://doi.org/10.1021/ac300373e, 2012.
Arnold, T., Manning, A. J., Kim, J., Li, S., Webster, H., Thomson, D., Mühle, J., Weiss, R. F., Park, S., and O'Doherty, S.: Inverse modelling of CF4 and NF3 emissions in East Asia, Atmos. Chem. Phys., 18, 13305–13320, https://doi.org/10.5194/acp-18-13305-2018, 2018.
Aydin, M., Saltzman, E. S., De Bruyn, W. J., Montzka, S. A., Butler, J. H., and Battle, M.: Atmospheric variability of methyl chloride during the last 300 years from an Antarctic ice core and firn air, Geophys. Res. Lett., 31, L02109, https://doi.org/10.1029/2003GL018750, 2004.
Battle, M., Bender, M., Sowers, T., Tans, P. P., Butler, J. H., Elkins, J. W., Ellis, J. T., Conway, T., Zhang, N., Lang, P., and Clarke, A. D.: Atmospheric gas concentrations over the past century measured in air from firn at the South Pole, Nature, 383, 231–235, 1996.
Bezuidenhoudt, A., Sonnendecker, P. W., and Crouse, P. L.: Temperature and pressure effects on the product distribution of PTFE pyrolysis by means of qualitative, in-line FTIR analysis, Polym. Degrad. Stab., 142, 79–88, https://doi.org/10.1016/j.polymdegradstab.2017.05.025, 2017.
Broyer, E., Bekker, A. Y., and Ritter, A. B.: Kinetics of the pyrolysis of chlorodifluoromethane, Ind. Eng. Chem. Res., 27, 208–211, https://doi.org/10.1021/ie00073a039, 1988.
Buizert, C., Martinerie, P., Petrenko, V. V., Severinghaus, J. P., Trudinger, C. M., Witrant, E., Rosen, J. L., Orsi, A. J., Rubino, M., Etheridge, D. M., Steele, L. P., Hogan, C., Laube, J. C., Sturges, W. T., Levchenko, V. A., Smith, A. M., Levin, I., Conway, T. J., Dlugokencky, E. J., Lang, P. M., Kawamura, K., Jenk, T. M., White, J. W. C., Sowers, T., Schwander, J., and Blunier, T.: Gas transport in firn: multiple-tracer characterisation and model intercomparison for NEEM, Northern Greenland, Atmos. Chem. Phys., 12, 4259–4277, https://doi.org/10.5194/acp-12-4259-2012, 2012.
Buznik, V. M.: Fluoropolymer chemistry in Russia: Current situation and prospects, Russ. J. Gen. Chem., 79, 520–526, https://doi.org/10.1134/s1070363209030335, 2009.
Cai, B., Liu, H., Kou, F., Yang, Y., Yao, B., Chen, X., Wong, D. S., Zhang, L., Li, J., Kuang, G., Chen, L., Zheng, J., Guan, D., and Shan, Y.: Estimating perfluorocarbon emission factors for industrial rare earth metal electrolysis, Resources, Conservation and Recycling, 136, 315–323, https://doi.org/10.1016/j.resconrec.2018.04.018, 2018.
Chinoy, P. B. and Sunavala, P. D.: Thermodynamics and kinetics for the manufacture of tetrafluoroethylene by the pyrolysis of chlorodifluoromethane, Ind. Eng. Chem. Res., 26, 1340–1344, https://doi.org/10.1021/ie00067a013, 1987.
Christophorou, L. G. and Olthoff, J. K.: Electron interactions with c-C4F8, J. Phys. Chem. Ref. Data, 30, 449–473, 2001.
Chung, M. O. and Bai, C.: Thermodynamic property variation of nonazeotropic refrigerant mixtures (NARMs) in the temperature gliding zone, Chem. Eng. Commun., 180, 1–17, https://doi.org/10.1080/00986440008912199, 2000.
Cunnold, D. M., Prinn, R. G., Rasmussen, R. A., Simmonds, P. G., Alyea, F. N., Cardelino, C. A., Crawford, A. J., Fraser, P. J., and Rosen, R. D.: The Atmospheric Lifetime Experiment, 3. Lifetime Methodology and Application to 3 Years of CFCl3 Data, J. Geophys. Res., 88, 8379–8400, 1983.
Cunnold, D. M., Weiss, R. F., Prinn, R. G., Hartley, D., Simmonds, P. G., Fraser, P. J., Miller, B., Alyea, F. N., and Porter, L.: GAGE/AGAGE measurements indicating reductions in global emissions of CCl3F and CCl2F2 in 1992–1994, J. Geophys. Res., 102, 1259–1269, 1997.
Cunnold, D. M., Steele, L. P., Fraser, P. J., Simmonds, P. G., Prinn, R. G., Weiss, R. F., Porter, L. W., O'Doherty, S., Langenfelds, R. L., Krummel, P. B., Wang, H. J., Emmons, L., Tie, X. X., and Dlugokencky, E. J.: In situ measurements of atmospheric methane at GAGE/AGAGE sites during 1985–2000 and resulting source inferences, J. Geophys. Res., 107, ACH-20-1–ACH-20-18, https://doi.org/10.1029/2001JD001226, 2002.
Dunse, B. L., Derek, N., Fraser, P. J., Krummel, P. B., and Steele, L. P.: Australian and global HFC, PFC, sulfur hexafluoride nitrogen trifluoride and sulfuryl fluoride emissions, Report prepared for Australian Government Department of the Environment and Energy, CSIRO Oceans and Atmosphere, Aspendale, Australia, iv, 33 pp., available at: http://www.environment.gov.au/protection/ozone/publications/csiro-report-australian-global-sgg (last access: 18 March 2019), 2018.
Engel, A., Rigby, M., Burkholder, J. B., Fernandez, R. P., Froidevaux, L., Hall, B. D., Hossaini, R., Saito, T., Vollmer, M. K., Yao, B., Altas, E., Bernath, P., Blake, D. R., Dutton, G., Krummel, P., Laube, J. C., Mahieu, E., Montzka, S. A., Mühle, J., Nedoluha, G., O'Doherty, S. J., Oram, D. E., Pfeilsticker, K., Prinn, R. G., Quack, B., Simpson, I. J., Weiss, R. F., Liang, Q., and Reimann, S.: Update on Ozone-Depleting Substances (ODSs) and Other Gases of Interest to the Montreal Protocol (Chapter 1), in: Scientific Assessment of Ozone Depletion: 2018, Global Ozone Research and Monitoring Project–Report No. 58, World Meteorological Organization, Geneva, Switzerland, 2018.
Fraser, P., Steele, P., and Cooksey, M.: PFC and Carbon Dioxide Emissions from an Australian Aluminium Smelter Using Time-Integrated Stack Sampling and GC-MS, GC-FID Analysis, in: Light Metals 2013, John Wiley & Sons, Hoboken, 871–876, 2013.
Fraser, P. J., Dunse, B. L., Manning, A. J., Walsh, S., Wang, R. H. J., Krummel, P. B., Steele, L. P., Porter, L. W., Allison, C., O'Doherty, S., Simmonds, P. G., Mühle, J., Weiss, R. F., and Prinn, R. G.: Australian carbon tetrachloride emissions in a global context, Environ. Chem., 11, 77–88, https://doi.org/10.1071/EN13171, 2014.
Fraser, P. J., Pearman, G. I., and Derek, N.: CSIRO Non-carbon Dioxide Greenhouse Gas Research. Part 1: 1975-90, Hist. Rec. Aust. Sci., 29, 1–13, https://doi.org/10.1071/HR17016, 2018.
Fuller, E. N., Schettler, P. D., and Giddings, J. C.: A new method for prediction of binary gas-phase diffusion coefficients, Ind. Eng. Chem., 58, 18–27, https://doi.org/10.1021/ie50677a007, 1966.
Ganesan, A. L., Rigby, M., Zammit-Mangion, A., Manning, A. J., Prinn, R. G., Fraser, P. J., Harth, C. M., Kim, K.-R., Krummel, P. B., Li, S., Mühle, J., O'Doherty, S. J., Park, S., Salameh, P. K., Steele, L. P., and Weiss, R. F.: Characterization of uncertainties in atmospheric trace gas inversions using hierarchical Bayesian methods, Atmos. Chem. Phys., 14, 3855–3864, https://doi.org/10.5194/acp-14-3855-2014, 2014.
Gangal, S. V. and Brothers, P. D.: Perfluorinated Polymers, in: Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Hoboken, 2015.
Harnisch, J.: Reactive Fluorine Compounds, in: Reactive Halogen Compounds in the Atmosphere Vol. 4 Part E, The Handbook of Environmental Chemistry, Springer, Berlin/Heidelberg, 81–111, 1999.
Harnisch, J.: Atmospheric perfluorocarbons: sources and concentrations, in: Non-CO2 greenhouse gases: scientific understanding, control and implementation, edited by: Ham, J. V., Baede, A., Meyer, L., and Ybema, R., Kluwer Academic Publishers, the Netherlands, 205–210, 2000.
Harnisch, J., Wing, I. S., Jacoby, H. D., and Prinn, R. G.: Primary Aluminum Production: Climate Policy, Emissions and Costs, Massachusetts Institute of Technology, Report #44, 1998.
Hastings, W. K.: Monte Carlo sampling methods using Markov chains and their applications, Biometrika, 57, 97–109, https://doi.org/10.1093/biomet/57.1.97, 1970.
Holliday, R. D. and Henry, J. L.: Anode polarization fluorocarbon formation in aluminum reduction cells, J. Ind. Eng. Chem., 51, 1289–1292, 1959.
Ivy, D. J., Arnold, T., Harth, C. M., Steele, L. P., Mühle, J., Rigby, M., Salameh, P. K., Leist, M., Krummel, P. B., Fraser, P. J., Weiss, R. F., and Prinn, R. G.: Atmospheric histories and growth trends of C4F10, C5F12, C6F14, C7F16 and C8F18, Atmos. Chem. Phys., 12, 4313–4325, https://doi.org/10.5194/acp-12-4313-2012, 2012.
Ji, L., Lu, S., Yang, J., Du, C., Chen, Z., Buekens, A., and Yan, J.: Municipal solid waste incineration in China and the issue of acidification: A review, Waste Manage. Res., 34, 280–297, https://doi.org/10.1177/0734242x16633776, 2016.
Jianming, S.: Synthesis of Hexafluoropropene by Co-pyrolysis Reaction of Tetrafluoroethylene with Octafluorocyclobutane, Masters Degree, Department of Chemical Engineering, Zhajiang University, Hangzhou, China, 2006.
Jones, A. R., Thomson, D. J., Hort, M., and Devenish, B.: The UK Met Office's next-generation atmospheric dispersion model, NAME III, in: Air Pollution Modelling and Its Application XVII, edited by: Borrego, C. and Norman, A.-L., Springer, Heidelberg, 580–589, 2007.
Kannan, G. K., Gupta, M., and Chandra Kapoor, J.: Estimation of gaseous products and particulate matter emission from garden biomass combustion in a simulation fire test chamber, Atmos. Environ., 39, 563–573, 2005.
Kass, W.: Tracing Technique in Geohydrology, CRC Press, New York, 1998.
Kim, J. H., Oh, C. H., Lee, N. E., and Yeom, G. Y.: Effect of N2O to C4F8∕O2 on global warming during silicon nitride plasma enhanced chemical vapor deposition (PECVD) chamber cleaning using a remote inductively coupled plasma source, Jpn. J. Appl. Phys. Part 2-Lett., 41, L1495–L1498, https://doi.org/10.1143/jjap.41.l1495, 2002.
Kokkoris, G., Goodyear, A., Cooke, M., and Gogolides, E.: A global model for C4F8 plasmas coupling gas phase and wall surface reaction kinetics, J. Phys. D-Appl. Phys., 41, 195211, https://doi.org/10.1088/0022-3727/41/19/195211, 2008.
Langenfelds, R. L., Krummel, P. B., Fraser, P. J., Steele, L. P., Ward, J., and Somerville, N. T.: Archiving of Cape Grim air, Baseline Atmospheric Program Australia 2009–2010, Melbourne, Australia, edited by: Derek, N., Krummel, P. B., and Cleland, S. J., 44–45, Australian Bureau of Meteorology and CSIRO Marine and Atmospheric Research, 2014.
Lewis Sr., R. J.: Food additives handbook, Chapman & Hall, New York, 1989.
Liu, X., Wang, J., Wang, Y., Zhang, Z., and Xiao, D.: Analysis of the insulation characteristics of c-C4F8∕CO2 gas mixtures by the Monte Carlo method, J. Phys. D-Appl. Phys., 41, 015206, https://doi.org/10.1088/0022-3727/41/1/015206, 2008.
Lovelock, J. E.: Atmospheric Fluorine Compounds as Indicators of Air Movements, Nature, 230, 5293, https://doi.org/10.1038/230379a0, 1971.
Lunt, M. F., Rigby, M., Ganesan, A. L., and Manning, A. J.: Estimation of trace gas fluxes with objectively determined basis functions using reversible-jump Markov chain Monte Carlo, Geosci. Model Dev., 9, 3213–3229, https://doi.org/10.5194/gmd-9-3213-2016, 2016.
Mackay, D., Shiu, W. Y., Ma, K. C., and Lee, S.: Handbook of Physical-Chemical Properties and Environmental Fate for Organic Chemicals, CRC Press, Boca Raton, 2006.
Maione, M., Giostra, U., Arduini, J., Furlani, F., Graziosi, F., Lo Vullo, E., and Bonasoni, P.: Ten years of continuous observations of stratospheric ozone depleting gases at Monte Cimone (Italy) – Comments on the effectiveness of the Montreal Protocol from a regional perspective, Sci. Total Environ., 445–446, 155–164, https://doi.org/10.1016/j.scitotenv.2012.12.056, 2013.
Matsunaga, N., Hori, M., and Nagashima, A.: Mutual diffusion coefficients of halogenated-hydrocarbon refrigerant-air systems, High Temp.-High Press, 25, 63–70, 1993.
Matsunaga, N., Hori, M., and Nagashima, A.: Measurements of the mutual diffusion coefficients of gases by the Taylor method, 7th report, measurements on the SF6-air, SF6-N2, SF6-O2, CFC12-N2, CFC12-O2, HCFC22-N2 and HCFC22-O2 systems, Trans. Jpn. Soc. Mech. Eng. B., 68, 550–555, 2002.
Matsunaga, N., Hori, M., and Nagashima, A.: Measurements of the mutual diffusion coefficients of carbon tetrafluoride and methyl bromide in air, nitrogen and oxygen, Proc. 26th Jpn. Symp. Therm. Props, 26, 499–501, 2005.
Metropolis, N., Rosenbluth, A. W., Rosenbluth, M. N., Teller, A. H., and Teller, E.: Equation of State Calculations by Fast Computing Machines, J. Chem. Phys., 21, 1087–1092, https://doi.org/10.1063/1.1699114, 1953.
Miller, B. R., Weiss, R. F., Salameh, P. K., Tanhua, T., Greally, B. R., Mühle, J., and Simmonds, P. G.: Medusa: A sample preconcentration and GC/MS detector system for in situ measurements of atmospheric trace halocarbons, hydrocarbons, and sulfur compounds, Anal. Chem., 80, 1536–1545, https://doi.org/10.1021/ac702084k, 2008.
Morisaki, S.: Simultaneous thermogravimetry-mass spectrometry and pyrolysis–gas chromatography of fluorocarbon polymers, Thermochim. Acta, 25, 171–183, 1978.
Morris, R. A., Miller, T. M., Viggiano, A. A., Paulson, J. F., Solomon, S., and Reid, G.: Effects of electron and ion reactions on atmospheric lifetimes of fully fluorinated compounds, J. Geophys. Res., 100, 1287–1294, 1995.
Mühle, J., Ganesan, A. L., Miller, B. R., Salameh, P. K., Harth, C. M., Greally, B. R., Rigby, M., Porter, L. W., Steele, L. P., Trudinger, C. M., Krummel, P. B., O'Doherty, S., Fraser, P. J., Simmonds, P. G., Prinn, R. G., and Weiss, R. F.: Perfluorocarbons in the global atmosphere: tetrafluoromethane, hexafluoroethane, and octafluoropropane, Atmos. Chem. Phys., 10, 5145–5164, https://doi.org/10.5194/acp-10-5145-2010, 2010.
Myhre, G., Shindell, D., Bréon, F.-M., Collins, W., Fuglestvedt, J., Huang, J., Koch, D., Lamarque, J.-F., Lee, D., Mendoza, B., Nakajima, T., Robock, A., Stephens, G., Takemura, T., and Zhang, H.: Anthropogenic and Natural Radiative Forcing, in: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Chapter 8, edited by: Stocker, T. F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S. K., Boschung, J., Nauels, A., Xia, Y., Bex, V., and Midgley, P. M., Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2013.
O'Doherty, S., Simmonds, P. G., Cunnold, D. M., Wang, H. J., Sturrock, G. A., Fraser, P. J., Ryall, D., Derwent, R. G., Weiss, R. F., Salameh, P., Miller, B. R., and Prinn, R. G.: In situ chloroform measurements at Advanced Global Atmospheric Gases Experiment atmospheric research stations from 1994 to 1998, J. Geophys. Res., 106, 20429–20444, 2001.
O'Doherty, S., Rigby, M., Mühle, J., Ivy, D. J., Miller, B. R., Young, D., Simmonds, P. G., Reimann, S., Vollmer, M. K., Krummel, P. B., Fraser, P. J., Steele, L. P., Dunse, B., Salameh, P. K., Harth, C. M., Arnold, T., Weiss, R. F., Kim, J., Park, S., Li, S., Lunder, C., Hermansen, O., Schmidbauer, N., Zhou, L. X., Yao, B., Wang, R. H. J., Manning, A. J., and Prinn, R. G.: Global emissions of HFC-143a (CH3CF3) and HFC-32 (CH2F2) from in situ and air archive atmospheric observations, Atmos. Chem. Phys., 14, 9249–9258, https://doi.org/10.5194/acp-14-9249-2014, 2014.
Oram, D.: Trends of long-lived anthropogenic halocarbons in the Southern Hemisphere and model calculations of global emissions, P.D. thesis, University of East Anglia, Norwich, UK, 1999.
Oram, D. E., Mani, F. S., Laube, J. C., Newland, M. J., Reeves, C. E., Sturges, W. T., Penkett, S. A., Brenninkmeijer, C. A. M., Röckmann, T., and Fraser, P. J.: Long-term tropospheric trend of octafluorocyclobutane (c-C4F8 or PFC-318), Atmos. Chem. Phys., 12, 261–269, https://doi.org/10.5194/acp-12-261-2012, 2012.
Prinn, R. G., Weiss, R. F., Fraser, P. J., Simmonds, P. G., Cunnold, D. M., Alyea, F. N., O'Doherty, S., Salameh, P., Miller, B. R., Huang, J., Wang, R. H. J., Hartley, D. E., Harth, C., Steele, L. P., Sturrock, G., Midgley, P. M., and McCulloch, A.: A history of chemically and radiatively important gases in air deduced from ALE/GAGE/AGAGE, J. Geophys. Res., 105, 17751–17792, https://doi.org/10.1029/2000jd900141, 2000.
Prinn, R. G., Huang, J., Weiss, R. F., Cunnold, D. M., Fraser, P. J., Simmonds, P. G., McCulloch, A., Harth, C., Salameh, P., O'Doherty, S., Wang, R. H. J., Porter, L., and Miller, B. R.: Evidence for substantial variations of atmospheric hydroxyl radicals in the past two decades, Science, 292, 1882–1888, 2001.
Prinn, R. G., Weiss, R. F., Arduini, J., Arnold, T., DeWitt, H. L., Fraser, P. J., Ganesan, A. L., Gasore, J., Harth, C. M., Hermansen, O., Kim, J., Krummel, P. B., Li, S., Loh, Z. M., Lunder, C. R., Maione, M., Manning, A. J., Miller, B. R., Mitrevski, B., Mühle, J., O'Doherty, S., Park, S., Reimann, S., Rigby, M., Saito, T., Salameh, P. K., Schmidt, R., Simmonds, P. G., Steele, L. P., Vollmer, M. K., Wang, R. H., Yao, B., Yokouchi, Y., Young, D., and Zhou, L.: History of chemically and radiatively important atmospheric gases from the Advanced Global Atmospheric Gases Experiment (AGAGE), Earth Syst. Sci. Data, 10, 985–1018, https://doi.org/10.5194/essd-10-985-2018, 2018.
Raju, R., Kudo, D., Kubo, Y., Inaba, T., and Shindo, H.: Warming potential reduction of C4F8 using inductively coupled plasma, Jpn. J. Appl. Phys. 1, 42, 280–285, https://doi.org/10.1143/jjap.42.280, 2003.
Ravishankara, A. R., Solomon, S., Turnipseed, A. A., and Warren, R. F.: Atmospheric lifetimes of long-lived halogenated species, Science, 259, 194–199, 1993.
Rigby, M., Ganesan, A. L., and Prinn, R. G.: Deriving emissions time series from sparse atmospheric mole fractions, J. Geophys. Res., 116, D08306, https://doi.org/10.1029/2010jd015401, 2011.
Rigby, M., Prinn, R. G., O'Doherty, S., Montzka, S. A., McCulloch, A., Harth, C. M., Mühle, J., Salameh, P. K., Weiss, R. F., Young, D., Simmonds, P. G., Hall, B. D., Dutton, G. S., Nance, D., Mondeel, D. J., Elkins, J. W., Krummel, P. B., Steele, L. P., and Fraser, P. J.: Re-evaluation of the lifetimes of the major CFCs and CH3CCl3 using atmospheric trends, Atmos. Chem. Phys., 13, 2691–2702, https://doi.org/10.5194/acp-13-2691-2013, 2013.
Rigby, M., Prinn, R. G., O'Doherty, S., Miller, B. R., Ivy, D., Mühle, J., Harth, C. M., Salameh, P. K., Arnold, T., Weiss, R. F., Krummel, P. B., Steele, L. P., Fraser, P. J., Young, D., and Simmonds, P. G.: Recent and future trends in synthetic greenhouse gas radiative forcing, Geophys. Res. Lett., 41, 2623–2630, https://doi.org/10.1002/2013gl059099, 2014.
Saito, T., Yokouchi, Y., Stohl, A., Taguchi, S., and Mukai, H.: Large emissions of perfluorocarbons in East Asia deduced from continuous atmospheric measurements, Environ. Sci. Technol., 44, 4089–4095, https://doi.org/10.1021/es1001488, 2010.
Sasaki, K., Kawai, Y., Suzuki, C., and Kadota, K.: Absolute density and reaction kinetics of fluorine atoms in high-density c-C4F8 plasmas, J. Appl. Phys., 83, 7482–7487, 1998.
Say, D., Ganesan, A. L., Lunt, M. F., Rigby, M., O'Doherty, S., Harth, C., Manning, A. J., Krummel, P. B., and Bauguitte, S.: Emissions of halocarbons from India inferred through atmospheric measurements, Atmos. Chem. Phys., 19, 9865–9885, https://doi.org/10.5194/acp-19-9865-2019, 2019.
Siegemund, G., Schwertfeger, W., Feiring, A., Smart, B., Behr, F., Vogel, H., McKusick, B., and Kirsch, P.: Fluorine Compounds, Organic, in: Ullmann's Encyclopedia of Industrial Chemistry, John Wiley & Sons, Hoboken, 2016.
Simmonds, P. G., Rigby, M., McCulloch, A., Vollmer, M. K., Henne, S., Mühle, J., O'Doherty, S., Manning, A. J., Krummel, P. B., Fraser, P. J., Young, D., Weiss, R. F., Salameh, P. K., Harth, C. M., Reimann, S., Trudinger, C. M., Steele, L. P., Wang, R. H. J., Ivy, D. J., Prinn, R. G., Mitrevski, B., and Etheridge, D. M.: Recent increases in the atmospheric growth rate and emissions of HFC-23 (CHF3) and the link to HCFC-22 (CHClF2) production, Atmos. Chem. Phys., 18, 4153–4169, https://doi.org/10.5194/acp-18-4153-2018, 2018.
Smith, A. M., Levchenko, V. A., Etheridge, D. M., Lowe, D. C., Hua, Q., Trudinger, C. M., Zoppi, U., and Elcheikh, A.: In search of in-situ radiocarbon in Law Dome ice and firn, Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms, 172, 610–622, https://doi.org/10.1016/S0168-583X(00)00280-9, 2000.
Sowers, T., Bernard, S., Aballain, O., Chappellaz, J., Barnola, J.-M., and Marik, T.: Records of the δ13C of atmospheric CH4 over the last 2 centuries as recorded in Antarctic snow and ice, Global Biogeochem. Cycles, 19, GB2002, https://doi.org/10.1029/2004GB002408, 2005.
Stanley, K. M., Grant, A., O'Doherty, S., Young, D., Manning, A. J., Stavert, A. R., Spain, T. G., Salameh, P. K., Harth, C. M., Simmonds, P. G., Sturges, W. T., Oram, D. E., and Derwent, R. G.: Greenhouse gas measurements from a UK network of tall towers: technical description and first results, Atmos. Meas. Tech., 11, 1437–1458, https://doi.org/10.5194/amt-11-1437-2018, 2018.
Stohl, A., Forster, C., Frank, A., Seibert, P., and Wotawa, G.: Technical note: The Lagrangian particle dispersion model FLEXPART version 6.2, Atmos. Chem. Phys., 5, 2461–2474, https://doi.org/10.5194/acp-5-2461-2005, 2005.
Stohl, A., Seibert, P., Arduini, J., Eckhardt, S., Fraser, P., Greally, B. R., Lunder, C., Maione, M., Mühle, J., O'Doherty, S., Prinn, R. G., Reimann, S., Saito, T., Schmidbauer, N., Simmonds, P. G., Vollmer, M. K., Weiss, R. F., and Yokouchi, Y.: An analytical inversion method for determining regional and global emissions of greenhouse gases: Sensitivity studies and application to halocarbons, Atmos. Chem. Phys., 9, 1597–1620, https://doi.org/10.5194/acp-9-1597-2009, 2009.
Sturges, W. T., Penkett, S. A., Lee, J. M., and Sturges, K. E.: Stratospheric distribution and lifetime of halogenated hydrocarbons; Final report to the European Commission, Contract EV5V-CT92-0078, Unversity of East Anglia, Norwich, 1995.
Sturges, W. T., Oram, D. E., Penkett, S. A., Fraser, P. J., and Engel, A.: Long-lived halogenated compounds in the stratosphere, in: Non-CO2 Greenhouse Gases: Scientific Understanding, Control, and Implementation, edited by: van Ham, J., Baede, A. P. M., Meyer, L. A., and Ybema, R., Kluwer Academic Publishers, the Netherlands, 239–240, 2000.
Sturrock, G. A., Etheridge, D. M., Trudinger, C. M., Fraser, P. J., and Smith, A. M.: Atmospheric histories of halocarbons from analysis of Antarctic firn air: Major Montreal Protocol species, J. Geophys. Res., 107, 4765, https://doi.org/10.1029/2002JD002548, 2002.
Tabereaux, A. T.: Anode Effects, PFCs, Global Warming, and the Aluminum-Industry, J. Miner. Met. Mater. Soc., 46, 30–34, 1994.
Travnicek, W.: A study of light halgenated hydrocarbons in stratospheric air samples using CG-MS, diploma thesis, Dept. Jülich, Fachhochschule Aachen, Germany, Aachen, 1998.
Trudinger, C. M., Enting, I. G., Etheridge, D. M., Francey, R. J., Levchenko, V. A., Steele, L. P., Raynaud, D., and Arnaud, L.: Modeling air movement and bubble trapping in firn, J. Geophys. Res., 102, 6747–6763, https://doi.org/10.1029/96jd03382, 1997.
Trudinger, C. M., Etheridge, D. M., Rayner, P. J., Enting, I. G., Sturrock, G. A., and Langenfelds, R. L.: Reconstructing atmospheric histories from measurements of air composition in firn, J. Geophys. Res., 107, 4780, https://doi.org/10.1029/2002JD002545, 2002.
Trudinger, C. M., Enting, I. G., Rayner, P. J., Etheridge, D. M., Buizert, C., Rubino, M., Krummel, P. B., and Blunier, T.: How well do different tracers constrain the firn diffusivity profile?, Atmos. Chem. Phys., 13, 1485–1510, https://doi.org/10.5194/acp-13-1485-2013, 2013.
Trudinger, C. M., Fraser, P. J., Etheridge, D. M., Sturges, W. T., Vollmer, M. K., Rigby, M., Martinerie, P., Mühle, J., Worton, D. R., Krummel, P. B., Steele, L. P., Miller, B. R., Laube, J., Mani, F. S., Rayner, P. J., Harth, C. M., Witrant, E., Blunier, T., Schwander, J., O'Doherty, S., and Battle, M.: Atmospheric abundance and global emissions of perfluorocarbons CF4, C2F6 and C3F8 since 1800 inferred from ice core, firn, air archive and in situ measurements, Atmos. Chem. Phys., 16, 11733–11754, https://doi.org/10.5194/acp-16-11733-2016, 2016.
Tsai, W. T.: Fate of Chloromethanes in the Atmospheric Environment: Implications for Human Health, Ozone Formation and Depletion, and Global Warming Impacts, Toxics, 5, 1–13, https://doi.org/10.3390/toxics5040023, 2017.
van der Walt, I. J., Neomagus, H. W. J. P., Nel, J. T., Bruinsma, O. S. L., and Crouse, P. L.: A kinetic expression for the pyrolytic decomposition of polytetrafluoroethylene, J. Fluor. Chem., 129, 314–318, https://doi.org/10.1016/j.jfluchem.2008.01.003, 2008.
Victor, D. G. and MacDonald, G. J.: A model for estimating future emissions of sulfur hexafluoride and perfluorocarbons, Clim. Change, 42, 633–662, 1999.
Vollmer, M. K., Mühle, J., Trudinger, C. M., Rigby, M., Montzka, S. A., Harth, C. M., Miller, B. R., Henne, S., Krummel, P. B., Hall, B. D., Young, D., Kim, J., Arduini, J., Wenger, A., Yao, B., Reimann, S., O'Doherty, S., Maione, M., Etheridge, D. M., Li, S., Verdonik, D. P., Park, S., Dutton, G., Steele, L. P., Lunder, C. R., Rhee, T. S., Hermansen, O., Schmidbauer, N., Wang, R. H. J., Hill, M., Salameh, P. K., Langenfelds, R. L., Zhou, L., Blunier, T., Schwander, J., Elkins, J. W., Butler, J. H., Simmonds, P. G., Weiss, R. F., Prinn, R. G., and Fraser, P. J. C. J. D.: Atmospheric histories and global emissions of halons H-1211 (CBrClF2), H-1301 (CBrF3), and H-2402 (CBrF2CBrF2), J. Geophys. Res., 121, 3663–3686, https://doi.org/10.1002/2015jd024488, 2016.
Vollmer, M. K., Young, D., Trudinger, C. M., Mühle, J., Henne, S., Rigby, M., Park, S., Li, S., Guillevic, M., Mitrevski, B., Harth, C. M., Miller, B. R., Reimann, S., Yao, B., Steele, L. P., Wyss, S. A., Lunder, C. R., Arduini, J., McCulloch, A., Wu, S., Rhee, T. S., Wang, R. H. J., Salameh, P. K., Hermansen, O., Hill, M., Langenfelds, R. L., Ivy, D., O'Doherty, S., Krummel, P. B., Maione, M., Etheridge, D. M., Zhou, L., Fraser, P. J., Prinn, R. G., Weiss, R. F., and Simmonds, P. G.: Atmospheric histories and emissions of chlorofluorocarbons CFC-13 (CClF3), ΣCFC-114 (C2Cl2F4), and CFC-115 (C2ClF5), Atmos. Chem. Phys., 18, 979–1002, https://doi.org/10.5194/acp-18-979-2018, 2018.
Vollmer, M. K., Bernard, F., Mitrevski, B., Steele, L. P., Trudinger, C. M., Reimann, S., Langenfelds, R. L., Krummel, P. B., Fraser, P. J., Etheridge, D. M., Curran, M. A. J., and Burkholder, J. B.: Abundances, emissions, and loss processes of the long-lived and potent greenhouse gas octafluorooxolane (octafluorotetrahydrofuran, c-C4F8O) in the atmosphere, Atmos. Chem. Phys., 19, 3481–3492, https://doi.org/10.5194/acp-19-3481-2019, 2019.
Walters, D. N., Williams, K. D., Boutle, I. A., Bushell, A. C., Edwards, J. M., Field, P. R., Lock, A. P., Morcrette, C. J., Stratton, R. A., Wilkinson, J. M., Willett, M. R., Bellouin, N., Bodas-Salcedo, A., Brooks, M. E., Copsey, D., Earnshaw, P. D., Hardiman, S. C., Harris, C. M., Levine, R. C., MacLachlan, C., Manners, J. C., Martin, G. M., Milton, S. F., Palmer, M. D., Roberts, M. J., Rodríguez, J. M., Tennant, W. J., and Vidale, P. L.: The Met Office Unified Model Global Atmosphere 4.0 and JULES Global Land 4.0 configurations, Geosci. Model Dev., 7, 361–386, https://doi.org/10.5194/gmd-7-361-2014, 2014.
Zhihong, W., Seidel, S., Kokorin, A., and Rand, S.: Chapter 3.6, PFC, HFC, SF6 emissions from semiconductor manufacturing, in: IPCC Good Practice Guidance and Uncertainty Management in National Greenhouse Gas Inventories, IGES, Hayama, Japan, 2001.