the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 13 Jul 2018
| 13 Jul 2018
Gas-phase composition and secondary organic aerosol formation from standard and particle filter-retrofitted gasoline direct injection vehicles investigated in a batch and flow reactor
Nivedita K. Kumar
Felix Klein
Pierre Comte
Deepika Bhattu
Josef Dommen
Emily A. Bruns
Doǧuşhan Kılıç
Imad El Haddad
Alejandro Keller
Jan Czerwinski
Norbert Heeb
Urs Baltensperger
Jay G. Slowik
André S. H. Prévôt
Gasoline direct injection (GDI) vehicles have recently been identified as a significant source of carbonaceous aerosol, of both primary and secondary origin. Here we investigated primary emissions and secondary organic aerosol (SOA) from four GDI vehicles, two of which were also retrofitted with a prototype gasoline particulate filter (GPF). We studied two driving test cycles under cold- and hot-engine conditions. Emissions were characterized by proton transfer reaction time-of-flight mass spectrometry (gaseous non-methane organic compounds, NMOCs), aerosol mass spectrometry (sub-micron non-refractory particles) and light attenuation measurements (equivalent black carbon (eBC) determination using Aethalometers) together with supporting instrumentation. Atmospheric processing was simulated using the PSI mobile smog chamber (SC) and the potential aerosol mass oxidation flow reactor (OFR). Overall, primary and secondary particulate matter (PM) and NMOC emissions were dominated by the engine cold start, i.e., before thermal activation of the catalytic after-treatment system. Trends in the SOA oxygen to carbon ratio (O : C) for OFR and SC were related to different OH exposures, but divergences in the H : C remained unexplained. SOA yields agreed within experimental variability between the two systems, with a tendency for higher values in the OFR than in the SC (or, vice versa, lower values in the SC). A few aromatic compounds dominated the NMOC emissions, primarily benzene, toluene, xylene isomers/ethylbenzene and C3-benzene. A significant fraction of the SOA was explained by those compounds, based on comparison of effective SOA yield curves with those of toluene, o-xylene and 1,2,4-trimethylbenzene determined in our OFR, as well as others from literature. Remaining discrepancies, which were smaller in the SC and larger in the OFR, were up to a factor of 2 and may have resulted from diverse reasons including unaccounted precursors and matrix effects. GPF retrofitting significantly reduced primary PM through removal of refractory eBC and partially removed the minor POA fraction. At cold-started conditions it did not affect hydrocarbon emission factors, relative chemical composition of NMOCs or SOA formation, and likewise SOA yields and bulk composition remained unaffected. GPF-induced effects at hot-engine conditions deserve attention in further studies.
- Article
(4359 KB) - Full-text XML
-
Supplement
(625 KB) - BibTeX
- EndNote





Vehicular emissions are a significant source of air pollution in many urban areas (Platt et al., 2014; Zotter et al., 2014; Bahreini et al., 2012; Borbon et al., 2013; May et al., 2014; Worton et al., 2014; Gentner et al., 2017). Depending on vehicle fleet technology, emissions may include fine particulate matter (PM), consisting mainly of sub-micron primary organic aerosol (POA) and black carbon (BC), and reactive gases such as nitrogen oxides (NOx) and organic compounds. Note that we refer to organic gas-phase compounds as non-methane organic compounds (NMOCs). Measurements by proton transfer reaction mass spectrometry (PTR-MS) are also referred to as NMOCs herein. Instead, when referring to measurements by flame ionization technique, we use the term non-methane hydrocarbons (NMHCs).
Human health is known to be impacted by NOx emissions, the associated ozone (O3) formation and fine PM emitted from combustion processes. Fine PM penetrates deep into the human body and can damage lung tissue (Kunzi et al., 2015) and likewise the brain (Calderon-Garciduenas and Villarreal-Rios, 2017). Therefore, numerous strategies have been developed to decrease PM and NOx emissions from on-road vehicles, including optimization of engine settings and implementation of after-treatment systems. Examples of such systems are oxidation catalysts that oxidize gas-phase pollutants (CO, NMOC), three-way catalysts (TWC) for gasoline on-road vehicles and selective catalytic reduction (SCR) systems for heavy-duty diesel engines and large diesel passenger cars, which convert NOx emissions to N2 and O2.
Historically, diesel-fueled vehicles have been recognized as a significant source of BC (Bond et al., 2004). Accordingly, the use of older generation diesel vehicles may be restricted in cities and catalyzed diesel particle filter (DPF) equipped diesel vehicles are subject to stringent primary PM limits. To achieve those, they are equipped with both diesel oxidation catalysts and DPF, which have trapping efficiencies for refractory material of up to 99 % (Gordon et al., 2013). Due to the regulatory attention and the improved after-treatment systems, diesel PM emissions from new generation vehicles have been greatly reduced, and fleet modernization can reduce their burden in the ambient air further. However, NOx emissions from diesel vehicles have not been addressed as successfully and remain a topic of debate (e.g., Barrett et al., 2015; Wang et al., 2016; di Rattalma and Perotti, 2017).
In contrast, gasoline light-duty vehicles have recently been engineered towards better fuel economy and reduced carbon dioxide (CO2) emissions to satisfy regulations aimed at mitigating climate change (Karjalainen et al., 2014). However, recent research indicates that some of the methods used to attain these emission goals, including smaller engines, leaner combustion and gasoline direct injection (GDI) systems mimicking the lower fuel consumption and decreased CO2 emission factors (EFs) of diesel vehicles, lead to an increase in the primary carbonaceous emissions, especially BC (Karjalainen et al., 2014; Zhu et al., 2016; Platt et al., 2017; Saliba et al., 2017; Zimmerman et al., 2016b). Consequently, modern gasoline light-duty vehicles have higher mass-based EFs of these pollutants than catalyzed-DPF equipped diesel vehicles (Platt et al., 2017). Additionally, they have been reported to emit ammonia (NH3) (Heeb et al., 2006; Suarez-Bertoa et al., 2014) formed on the gasoline TWC. These emissions are released predominantly at engine start-up, when catalytic after-treatment systems are still cold, and during acceleration and deceleration (Platt et al., 2017; Gentner et al., 2017).
Automobile manufacturers have recently considered equipping gasoline light-duty vehicles with gasoline particulate filters (GPFs) in the light of increasingly stringent legislations regarding PM abatement. First results are promising (Chan et al., 2014; Demuynck, 2017; Czerwinski et al., 2017). Although GPFs are likely to be similarly effective as DPFs in reducing primary PM such as POA and BC, recent research indicates that the dominant fraction of the total PM from modern gasoline vehicles is secondary (Platt et al., 2013, 2017; Nordin et al., 2013; Gordon et al., 2014a, b; Gentner et al., 2017). Dominant secondary species include secondary organic aerosol (SOA) (Hallquist et al., 2009) and ammonium nitrate (NH4NO3), which are formed by the reaction of emitted NMOCs and NOx in presence of NH3, respectively, with atmospheric oxidants such as hydroxyl radicals (OH). The gaseous precursors leading to secondary aerosol are unlikely to be removed by GPF systems alone. Laboratory results of the GPF effect on NMOC emissions and the associated SOA formation are, however, lacking.
Detailed investigations of SOA formation are typically performed in smog chambers (SC), where the emitted gases are oxidized in batch-style experiments lasting several hours under close-to-tropospheric conditions. The poor time resolution of such experiments prevents efficient study of SOA formation as a function of driving conditions (e.g., engine load or catalyst temperature), which as noted above is a critical consideration for gasoline vehicles. In contrast, oxidation flow reactors (OFR) (Kang et al., 2007; Li et al., 2015) are based on flow-through systems, allowing for investigation of SOA formation from time-varying emissions. They utilize higher-than-ambient oxidant concentrations to simulate hours to days of atmospheric aging in only a few minutes of experimental time. Several studies have attempted the quantitative application of OFR systems to complex combustion emissions (Zhao et al., 2018; Karjalainen et al., 2016; Bruns et al., 2015; Tkacik et al., 2014; Ortega et al., 2013). An in-depth analysis of SC and OFR application to GDI exhaust, however, remains missing, and the OFR oxidation conditions (e.g., high oxidant concentrations, short-wavelength light spectrum, high wall surface-to-volume ratios) require further investigation (Lambe et al., 2011, 2015, 2017; Peng et al., 2015, 2016; Li et al., 2015; Palm et al., 2016).
Despite recent studies of SOA from gasoline vehicle exhaust (Platt et al., 2013, 2017; Gordon et al., 2014a, b; Nordin et al., 2013; Zhao et al., 2017, 2018), the formation processes, the role of relevant precursors and their SOA yields remain a subject of debate, and SOA data from the European GDI fleet are scarce. A wide range of ratios of secondary-to-primary OA (SOA ∕ POA) and SOA yields (mass of SOA formed per organic vapors reacted) has been reported while using standardized and repeatable testing procedures (Jathar et al., 2014; Gentner et al., 2017). This is in part due to the high uncertainty related to the effect of experimental considerations, including NMOC levels, NO concentrations, OH exposure, particle and vapor wall losses and emissions sampling. Moreover, the previously applied techniques (such as offline gas-chromatography MS and FID analysis of the total hydrocarbons (THC), or quadrupole PTR-MS (Q-PTR-MS) (de Gouw et al., 2003; Lindinger and Jordan, 1998), which allowed only for online monitoring of selected compounds with no significant interferences at the same integer m∕z) show limitations when studying complex combustion emissions. Recently, Zhao et al. (2016) suggested that the precursors are dominantly volatile organic compounds with a saturation concentration, C*, above 106 µg m−3. This should allow for investigation with modern online instrumentation, such as the high-resolution time-of-flight PTR-MS (PTR-ToF-MS).
Here, we investigated primary NMOC, POA, equivalent black carbon (eBC) emissions and SOA formation from Euro 4 and Euro 5 GDI vehicle exhaust, including vehicles retrofitted with prototype GPFs. Vehicles were tested on a chassis dynamometer during a modern regulatory driving cycle (worldwide light-duty test cycle, WLTC class 3) and an older low-load European driving cycle (EDC), under both cold- and hot-engine conditions. We studied SOA formation through batch-style aging in the PSI mobile SC (Platt et al., 2013) and the potential aerosol mass OFR, (Bruns et al., 2015; Lambe et al., 2011, 2015), applying the latter for both batch-style and time-resolved analysis. Relevant SOA precursors were characterized using a PTR-ToF-MS, and their photochemical processing was related to SOA formation, where SOA mass and its bulk chemical composition was derived from HR-ToF-AMS measurements.
Two experimental sets were conducted (set I in 2014, set II in 2015). In addition, selected SOA precursors (toluene, o-xylene and 1,2,4-trimethylbenzene (TMB)) were separately injected into the OFR for comparison with the vehicle data (experiments conducted in 2016). In the following we describe vehicle testing (Sect. 2.1), photochemistry experiments (Sect. 2.2) and mass spectrometric instrumentation (Sect. 2.3).
2.1 Vehicle testing
Vehicles were operated on a chassis dynamometer at the Laboratories for IC-Engines and Exhaust Emission Control of the Berne University of Applied Sciences in Biel (Switzerland) that includes a roller dynamometer (Schenck 500 GS60), a driver conductor system (Tornado, version 3.3), a CVS-dilution system (Horiba CVS-9500T with Roots blower) and an automatic air conditioning in the hall (intake and dilution air) to maintain a temperature of 20–30 ∘C and an absolute humidity of 5.5–12.2 g kg−1. The driving resistances of the test bench and the braking resistances were set according to legal prescriptions without elevation change. This equipment fulfilled the requirements of the Swiss and European exhaust gas legislation. The dilution ratio in the CVS-dilution tunnel was variable and assessed by means of the CO2 analysis; the range was from 8, during high engines loads, to 30–40, at idle conditions. Gaseous components were monitored with the exhaust gas measuring system Horiba MEXA-9400H, including measurements of CO and CO2 by infrared analyzers (IR) and hydrocarbons by flame ionization detector (FID) for THC and NMHC measurements. Further instrumentation is listed in Sect. S2 in the Supplement.
2.1.1 Vehicles, GPFs and fuels
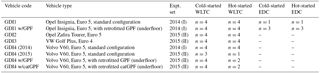
Tables 1 and S1 list the tested vehicles. In 2014, we tested a GDI Opel Insignia (denoted GDI1) and a Volvo V60 (GDI4). GDI1 was studied in standard configuration, and also retrofitted with a prototype GPF (cordierite, porosity 50 %, pore size 19 µm, 2000 cells per square inch). The GPF was installed “underfloor”, ca. 60 cm downstream of the original TWC, and replaced the muffler (Fig. S1 in the Supplement). Filtration quality at this configuration was equivalent to the best available technology for DPFs (personal communication with the manufacturer; particle number reductions were further assessed in Czerwinski et al., 2017, and yielded a particle count filtration efficiency (PCFE) ≥ 98 %). In 2015, we tested two additional GDI vehicles (GDI2, GDI3) in standard configuration. We also repeated tests with GDI4 in standard configuration and retrofitted with two GPFs: (a) the previously tested GPF (as above) and (b) a Pd ∕ Rh catalytically coated GPF (denoted catGPF). Retrofitting was again performed in the form of an underfloor modification replacing the muffler ca. 60 cm downstream the original TWC. The PCFE was ≥ 86 %. The primary purpose of the catalytically active coating was constant GPF self-cleaning of deposited carbonaceous material according to personal communication with the manufacturer. In future applications, such catalytic coatings might replace the existing TWC or, more specifically, the TWC could be combined with a GPF in one system. All vehicles were fueled with gasoline from the Swiss market, RON 95, according to SN EN228. It contained 35 % aromatic hydrocarbons (ArHC), < 1 % alkenes, 5 % methyl tert-butyl ether (MTBE) (in 2014, ∼ 8 % in 2015) added as anti-knocking agent and < 0.5 % ethanol, all on a volumetric basis.
Table 1Vehicles (details in Table S1) and tests (n gives the number of driving tests conducted; EDC tests were only conducted with GDI1 and GDI1 w/GPF).
2.1.2 Test cycles
We used dynamic driving cycles: the worldwide light-duty test cycle (WLTC-class 3) and the common, but nowadays considered less representative, EDC. Figures 1 and S2 provide the speed profiles. While the EDC is characterized by two phases, the urban and extra-urban phases of highly repetitive characteristics, and lasts 20 min, the WLTC has four phases at different speed levels, referred to as phases (Ph) 1–4 (i.e., low, medium, high, extra-high speed) and contains patterns of disruptive acceleration and deceleration. It lasts 30 min. Engines were started either after a soaking time of at least 6 h at test bench temperature (referred to as “cold-started”) or after warming the engine and after-treatment system by driving for 3 min at steady state (80 km h−1, “hot-started”). Tests are referred to as cold-started WLTC (cW), hot-started WLTC (hW), cold-started EDC (cE) and hot-started EDC (hE) throughout the paper.
2.2 Non-regulatory measurements and photochemistry experiments
In parallel to the CVS measurements, emissions were sampled from the tailpipe using either one or two Dekati ejector dilutors in series for characterization by non-regulatory equipment and photochemistry experiments. Figure 1 gives a scheme of the setup. Sampling was performed as reported earlier in Platt et al. (2013, 2017). It demonstrated good agreement of batch-sampled emissions with online measurements of gaseous pollutants at the tailpipe (Platt et al., 2013) and also gravimetric PM samples from the CVS (Platt et al., 2017). A likewise comparison of our PM measurements is provided in Fig. S16. Tubing material, length, temperature and flow rates are specified in the Sect. S3 in the Supplement. Clean air to operate the non-CVS sampling and dilution system, SC and OFR, was provided by a compressor (Atlas Copco SF 1 oil-free scroll compressor with 270 L container, Atlas Copco AG, Switzerland) combined with an air purifier (AADCO 250 series, AADCO Instruments, Inc., USA). Clean air specifications can be found in Platt et al. (2013). Along with measurements of CO2, CO and CH4 (CRDS, Picarro), THC, CH4 and NMHC (FID, Horiba), NO, NO2 and O3 particle-phase instruments (CPC and SMPS for particle number and size measurements, and seven-wavelength Aethalometers for eBC determination (Drinovec et al., 2015) Aerosol d.o.o), we deployed high-resolution time-of-flight mass spectrometers to investigate the chemical composition of the fresh and aged exhaust. Mass spectrometric instrumentation is described in Sect. 2.3 and all instruments are listed in Tables S2–S3.
2.2.1 Experimental procedure
Experiments were conducted in three configurations:
-
time-resolved measurements of primary emissions and time-resolved aging in the OFR during dynamic driving cycles, denoted “OFR online”;
-
OFR photochemical aging from SC batch samples which were collected over a driving cycle or phases there, denoted “OFR-from-SC”;
-
SC photochemical aging of the previously mentioned SC batch sample.
At the start of each experiment the cleaned SC was filled to approximately two-thirds full with humidified air, with the remaining volume available for sample injection. Diluted emissions from the cold-started driving cycle were then sampled into the SC for a later photochemical batch experiment. The batch sampling was conducted over either the full cycle (cW and cE), the first phase (Ph 1, cW) or the aggregated second through fourth phases (Ph 2–4, cW). After sample injection, the chamber volume was filled up to its maximum with clean air, and the relative humidity was adjusted to 50 %. To quantify OH exposure during the later photochemical experiments, 1 µL of nine-times deuterated BuOH (BuOH-D9, purchased from Cambridge Isotope Laboratories) was added to the sample (Barmet et al., 2012).
In parallel to SC sampling, diluted emissions from the cold-started tests were sampled online during the test bench driving cycle and characterized in real time, either fresh (“primary”) or OFR photochemically aged (“secondary”). Once the first driving test was completed and the primary emissions were characterized in the SC batch, a hot-started vehicle test was performed. For this purpose, the vehicle was operated for 3 min at 80 km h−1 steady-state driving prior to the test. Emissions of the hot-started cycle were characterized in real-time fresh or OFR aged. No sampling of hot-started cycle emissions into the SC was performed.
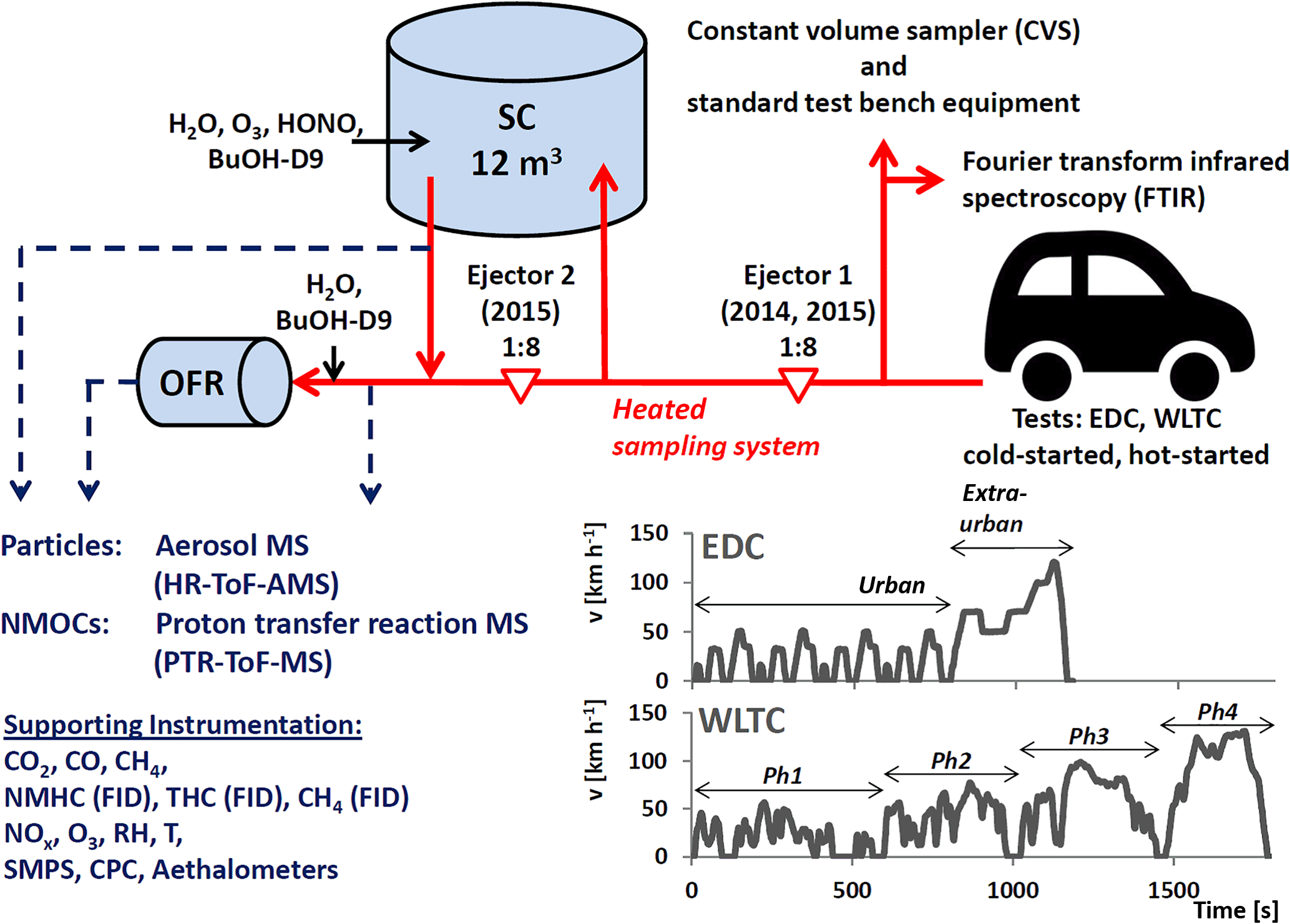
Figure 1Schematic (not to scale) of the experimental setup. Vehicles were driven over regulatory driving cycles (EDC and WLTC, for which speed profiles are shown in the figure) on a chassis dynamometer test bench. Emissions were sampled through a heated dilution and sampling system using one or two ejector dilutors into the PSI mobile SC (Platt et al., 2013) and the potential aerosol mass oxidation flow reactor (OFR) (Bruns et al., 2015). Instrumentation for characterization of fresh and photochemically aged emissions is listed. The raw exhaust was also sampled at the tailpipe using standard test bench equipment to monitor regulatory species (diluted in a constant volume sampler, CVS) and unregulated emissions (with Fourier-transformed infrared spectroscopy).
Once both driving tests were completed, the emissions previously collected in the SC were characterized, and when the monitored parameters and BuOH-D9 signal stabilized and indicated a well-mixed chamber, primary emissions were sampled from the SC into the OFR for photochemical aging (“OFR-from-SC” sampling, also referred to as “batch OFR” herein). The OFR was operated at varied OH exposures determined by UV lamp intensity (100, 70, 50 %). Finally, UV “on” measurements were followed by a UV “off” (OFR dark) period. Once OFR-from-SC sampling was completed, O3 was injected into the SC to titrate NO to NO2. Nitrous acid (HONO), used as an OH precursor in the SC, was injected continuously for the remainder of the experiment and photochemistry was initiated by illuminating the SC with UV lights for a period of 2 h. The temperature around the SC was initially 23±2 ∘C and reached 26±2 ∘C with UV lights on. The OFR likely also exhibited slightly higher than ambient temperatures close to the UV sources due to heating from the lamps. Background measurements were conducted before every experiment in SC and OFR (see Sects. 2.2.2 and 2.2.3).
In addition to GDI exhaust experiments, toluene, o-xylene and 1,2,4-TMB provided via a liquid injection system were aged in the OFR as reference measurements in separate experiments.
2.2.2 PSI mobile smog chamber
The SC described by Platt et al. (2013) is an approximately 12 m3, 125 µm thick collapsible Teflon bag (DuPont Teflon fluorocarbon film (FEP), type 500A, Foiltec GmbH, Germany) suspended from a mobile aluminum frame ( m, ) with a battery of 40×100 W UV lights (Cleo Performance solarium lamps, Philips). It is equipped with an injection system for purified air, water vapor and gases. OH radicals used as the primary oxidant are generated by photolysis of HONO (Platt et al., 2013; Taira and Kanda, 1990). During photochemistry, in situ formation of O3 resulted in an average OH ∕ O3 ratio of ; OH concentration and exposure are provided in the results section. The SC was cleaned prior to each experiment by filling with humidified air and O3, irradiating with UV light for at least 1 h, followed by flushing with dry, pure air for at least 10 h. Background measurements of the clean SC were conducted prior to each experiment with UV lights off. Background was insignificant compared to our measurements, except when stated otherwise. Photochemistry control experiments were conducted regularly to estimate the contribution of the SC background to SOA formation; these experiments were conducted after the standard cleaning procedure. Instead of vehicle exhaust, pure air was used as a sample and ammonium sulfate (50 µg m−3) injected as seed. Other photochemistry experimental procedures were in line with the typical vehicle experiments. We found a SOA background < 1 µg m−3. This was below the SOA concentrations formed during vehicle exhaust aging; see concentration levels as reported in Tables S4–S7. Concentration levels in the SC, which were a result of our sampling and dilution strategy, were representative for urban ambient conditions.
2.2.3 Oxidation flow reactor
Experiments herein utilize the potential aerosol mass OFR, of which several different configurations exist (Bruns et al., 2015; Lambe et al., 2011, 2015; Kang et al., 2007). Our OFR was previously described by Bruns et al. (2015) and consists of a 0.015 m3 cylindrical glass chamber (0.46 m length, 0.22 m diameter) containing two low-pressure mercury lamps, each with discrete emission lines at 185 and 254 nm (BHK Inc.) (Li et al., 2015; Peng et al., 2015, 2016). The lamps were cooled by a constant flow of air. The incoming reactant flow was dispersed by a perforated mesh screen at the inlet flange. In our experiments, the flow through the OFR was regulated by the flow pulled by instruments and pumps behind the reactor and was set to ∼ 8–9 L min−1. This corresponds to a plug flow residence time of 90–100 s. A fraction of the total flow (0.5–1 L min−1) was sampled behind a second perforated mesh and discarded to limit wall effects. The OFR was equipped with an injection system for water vapor (a Nafion humidifier) and organic compounds (BuOH-D9 as an OH tracer and toluene, o-xylene and 1,2,4-TMB purchased from Sigma-Aldrich (p.a.) for precursor tests). Figure S3 provides a scheme. OH radicals in the OFR were produced by photolysis of water vapor at 185 nm and by production of atomic oxygen in excited state O(1D) from photolysis of ozone (O3) at 254 nm, which can react with H2O to form OH. O3 itself was produced by reaction of atomic oxygen in ground state, O(3P), with O2. O(3P) in turn was formed by photolysis of O2 at 185 nm. Lamp power can be regulated between 0 and 100 %, with lower intensities lowering both O3 and OH production. The ratio of OH ∕ O3 remained relatively constant at our test points: (1.4–2.6) × 10−5 at 100 %, (1.9–3.0) × 10−5 at 70 % and (1.7–2.6) × 10−5 at 50 %. OH concentration and exposure are provided in the results section. During “online” (time-resolved) operation, the diluted exhaust (one or two ejector dilutors, each at a dilution ratio of 1 : 8) was mixed with humidified air up to 50 % of the total volume flow through the reactor. For OFR-from-SC experiments instead, no separate addition of water vapor or BuOH-D9 was required. The OFR was cleaned prior to each experiment by flushing with humidified, pure air, while keeping UV lights on for at least 10 min. Background levels were < 2 µg m−3 SOA before OFR-from-SC experiments (when sampling from cleaned SC) and < 10 µg m−3 when sampling diluted (1 : 8) test bench room air prior to online experiments.
2.2.4 Particle losses in SC and OFR
Loss of particulate (and gaseous) material to reactor walls can cause significant uncertainties in simulations of atmospheric aging (Zhang et al., 2014; Lambe et al., 2011; McMurry and Grosjean, 1985). The main losses of particles are due to (1) diffusion, (2) electrostatic deposition and (3) gravitational settling, which are in turn affected by temperature changes due to the UV lights.
Those losses were accounted for in our SC experiments using the method described in Weitkamp et al. (2007) and Hildebrandt et al. (2009). This addresses all effects, including the aforementioned temperature effects, simultaneously. The suspended OA concentration, COA, suspended, was consequently corrected to yield COA, wlc following Eq. (1) from Hildebrandt et al. (2009). Particulate wall loss rates, kw, were determined from an exponential fit of the time-dependent decrease in eBC mass determined from optical absorption at λ= 950 nm. When eBC was below the instrumental detection limit (e.g., for experiments with retrofitted GPF), an average based on the other experiments was applied (kw=5.6 × 10−5 s−1). Diffusional losses of particles vary with particle size (McMurry and Grosjean, 1985). Our correction implicitly assumed internally mixed OA–eBC particles and did not account separately for size-dependent effects.
A comparison of eBC mass up- and downstream the OFR indicated no significant losses of particulates during UV on or UV off periods; the experimentally determined transmission was equal to 1. Consequently no further correction was applied. Particle wall losses in the OFR have been quantified previously by Lambe et al. (2011), who reported at least 80 % transmission efficiency for particles of mobility diameter (dm) > 150 nm. The particles measured downstream the OFR in our study had a median vacuum aerodynamic diameter (dva) between 200 and 400 nm (HR-ToF-AMS-based size distributions in Fig. S10). This corresponded to dm > 150 nm assuming spherical particles and an OA density of 1.2 g cm−3 (Turpin and Lim, 2001). Particle size distributions in this range supported our experimentally determined transmission equal to 1.
2.2.5 Vapor losses in SC and OFR (on walls and through other non-OH processes)
Low-volatility vapors, especially semi-volatile (SVOC) and low-volatility organic compounds (LVOC), are prone to losses on clean reactor walls and deposited OA particles, which compete with partitioning to suspended OA particles. Numerous publications discussed potential SVOC and LVOC wall losses in SC systems recently (e.g., Krechmer et al., 2016; Ye et al., 2016; Zhang et al., 2014; Hildebrandt et al., 2009). They highlighted that these losses may result in underpredictions of SOA yields. However, a robust strategy for their determination and correction remains challenging (Krechmer et al., 2016). In our previous work, we estimated that vapor wall losses may cause SOA yields to be underestimated for the specific SC used herein by a factor of 1.5–2 (assessed based on gasoline vehicle exhaust; see Platt et al., 2017). This is in line with suggestions by others (e.g., a factor of 1.1.–4.2 reported by Zhang et al., 2014, and 1.1–6 reported by La et al., 2016). Hence, data correction would increase our SC SOA yields on average by a factor of 1.5–2.
Palm et al. (2016) recently estimated LVOC losses in the OFR and described them as a result of losses to walls, losses due to insufficient residence time for partitioning to the particle phase (i.e., vapors exit the OFR before they condense) and losses due to fragmentation upon multiple OH reactions prior to vapor condensation on suspended OA. We tested the loss rate of vapors in our OFR for batch mode operation. Given the high SOA concentration and hence high available particle surface ((1−5) × 109 nm2 cm−3 based on the SMPS size distribution of SOA), less than 20 % of the formed LVOC was estimated to be lost to the reactor walls using the Palm et al. (2016) model. Data correction would increase our OFR SOA yields by a factor of 1.25 on average.
Non-OH reaction processes in the OFR can be another pathway by which SOA precursors (vapors) are lost. These processes have been parameterized by Peng et al. (2016) as a function of residence time, photon flux or O3 measurements, water vapor availability, and external OH reactivity (OHRext), which is defined as the product of the available OH-reactive material and its respective OH rate constant. Photons (185, 254 nm), oxygen allotropes (excited oxygen atoms (O(1D)), ground state oxygen atoms (O(3P)) and ozone (O3)) were identified as relevant loss processes to precursor molecules, dependent on their chemical identity. To estimate their contribution vs. OH reactions, we applied the Peng et al. (2016) model. The results and implications of photon-induced effects on SOA formation or destruction are discussed in the Sect. S4 in the Supplement. In brief, for OFR-from-SC experiments, we predicted non-OH loss processes of SOA precursors to yield up to 25 % for benzene and 10 % for toluene, initiated by photons. For time-resolved OFR experiments, the model predicted more significant losses at low dilution ratios (one ejector dilutor, which applies to experiments from 2014) and smaller influences for experiments conducted with double dilution (two ejector dilutors, which applies to experiments from 2015). This is due to the relatively higher [OH] at lower OHRext. Time-resolved OFR experiments from 2014 were further impacted by OH suppression and relatively higher NOx levels. For this reason we did not use them quantitatively within this publication (see Sects. 3.3 and S4).
2.2.6 SOA yields
Our SOA yield analysis is based on SC and OFR-from-SC experiments with GDI1-3 when tested over the full cycle or Ph1 only. An effective SOA yield (Ye) was calculated as the ratio of the formed SOA mass to the reacted SOA-forming species i (in Δ µg m−3, Eq. 2). We took into account all our identified SOA precursors, i.e., the eight dominant ArHC presented in Fig. 4d, neglecting non-reactive and non-SOA-forming precursors. Thereby, we assumed that all relevant SOA precursors were measured.
SOA yields are presented as a function of the suspended (i.e., non particle wall loss corrected) organic aerosol mass (POA + SOA), for consistency with the wall loss correction method described above (i.e., neglecting vapor–wall interactions). In practice, this has little effect on the obtained SOA yield curves, as particle wall losses were limited due to the short experiment time (2 h). As the yield could be calculated for each point in time since initiation of photochemistry, values as a function of OH exposure and also as a function of suspended OA were derived.
2.2.7 OH exposure estimation
The time-integrated OH exposure (molec cm−3 s), defined as the integrated OH concentration over the reaction time (t), was calculated from the decay of BuOH-D9 (Barmet et al., 2012). The obtained OH exposure was related to an approximate ambient aging time by assuming a mean atmospheric [OH] of 1 × 106 molec cm−3 (global 24 h average; Finlayson-Pitts and Pitts, 2000). We also predicted OH concentration and exposure using the OFR model and OH exposure estimation from Li et al. (2015) and Peng et al. (2016). The tracer-based OH exposure was generally in good agreement with the model results, except at the highest OH exposures where the tracer method was on average a factor of 3 higher (Sect. S4 in the Supplement). Tracer-based OH exposures were used throughout our analysis, as these measurements are specific to our experiments.
2.3 Mass spectrometric instrumentation
2.3.1 PTR-ToF-MS
A PTR-ToF-MS (PTR-TOF-8000, Ionicon Analytik GmbH, Innsbruck, Austria; Jordan et al., 2009; Graus et al., 2010) was used to study gaseous NMOC in fresh and aged emissions. We used hydronium ions (H3O+) as the primary reagent. Water clusters (H3O)(H2O)+ were below 5 % of the H3O+ ion and not considered further. Detected compounds included ArHC, alkanes (> C10) and alkenes (> C2), as well as oxygenated compounds and thus many molecules expected in GDI vehicle exhaust (Gueneron et al., 2015; Schauer et al., 2002). For set I (2014), the PTR-ToF-MS operated at a drift voltage of 545 V, a chamber temperature of 90 ∘C, a drift pressure of 2.2 mbar and a resulting reduced electric field (E∕N) of 140 Td. In set II (2015) and for single precursor experiments (2016), we used 545 V, 60 ∘C and 2.1–2.2 mbar, respectively, resulting in an E∕N of 130 Td. The mass resolution, mass accuracy and relative transmission efficiency (De Gouw and Warneke, 2007; Müller et al., 2014) were routinely verified using a 12-compound gas standard (Carbagas; protonated integer m∕z 45 to 181, containing alcohols, carbonyls, alkenes, aromatic hydrocarbons and terpenes). Further, we used an internal calibrant (diiodobenzene, C6H4I2, protonated integer m∕z 331) to support mass calibration at higher m∕z.
Data were analyzed using the Tofware post-processing software (version 2.4.2, TOFWERK AG, Thun, Switzerland; PTR module as distributed by Ionicon Analytik GmbH, Innsbruck, Austria), running in the Igor Pro 6.3 environment (Wavemetrics Inc., Lake Oswego, OR, USA). In the absence of fragmentation, ions are observed at the m∕z corresponding to the neutral parent molecule shifted by the mass of one proton (denoted ). The exact mass was used to determine the elemental composition and combined with previous reports of compounds identified in emissions (Schauer et al., 1999, 2002; Gueneron et al., 2015; Erickson et al., 2014) to propose likely molecular structures. NMOC concentrations were derived from the H3O+ normalized ion signal of , the appropriate reaction rate constant towards H3O+( from Cappellin et al. (2010, 2012), and the residence time in the drift tube, following standard procedures. While ideally the molecular sum formula can be approximated by the exact mass of , isomers, such as o-, p- and m-xylenes and ethylbenzene, cannot be resolved and is uncertain. When the information was missing, we used the collisional rate constant ( cm3 s−1). Although protonation with H3O+ is typically soft, fragmentation may occur for aldehydes, alcohols, alkanes, alkenes and substituted aromatics, with the non-oxygen-containing species being of particular importance herein (Gueneron et al., 2015; Erickson et al., 2014; Buhr et al., 2002). Fragments constituted a small fraction of the total signal in our analysis (see results). No corrections were applied.
2.3.2 HR-ToF-AMS
Quantitative, size-resolved mass spectra of the non-refractory sub-micron particle composition were obtained using a high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS, Aerodyne, DeCarlo et al., 2006), equipped with a PM1 aerodynamic lens. All data presented herein are open minus closed signals derived from high-resolution analysis fitting procedures (SQUIRREL1.51H, PIKA 1.10H), running in the Igor Pro 6.3 environment (Wavemetrics Inc., Lake Oswego, OR, USA). Following standard procedures (Canagaratna et al., 2007), the instrument ionization efficiency and particle size measurement were calibrated using size-selected NH4NO3 particles and polystyrene latex spheres, respectively. A relative ionization efficiency of 1.4 for organic material was applied. We used a collection efficiency of 1 because, through photochemistry, significant amounts of NH4NO3 were formed, and under our (NH4)2SO4-free conditions our aerosol mixture is not expected to bounce significantly. No corrections for lens transmission were performed; particle time-of-flight distributions are provided in Fig. S10. HR-ToF-AMS data were corrected for background gas-phase CO2 by subtracting the CO2 signal measured in a particle-free sample. The interaction of inorganic salts with pre-deposited carbon on the tungsten vaporizer can lead to a signal in the open minus closed HR-ToF-AMS mass spectra (Pieber et al., 2016). Photochemical aging resulted in significant NH4NO3 formation, reaching NO3 ∕ OA ratios of roughly 5. A interference equivalent to 3.5 % of the NO3 concentration was determined by calibration (Fig. S4) and corrected according to Pieber et al. (2016).
3.1 Pollutant EFs as function of vehicle technology and driving cycle
Figure 2 summarizes EFs across all vehicles and conditions tested. We investigated NMHC, THC, primary PM (eBC, POA) and SOA. A detailed discussion on emissions of CO, NOx, particle number and genotoxic polycyclic aromatic hydrocarbons from cold- vs. hot-started cycle-driven GDI vehicles in standard configuration is provided in Muñoz et al. (2018).
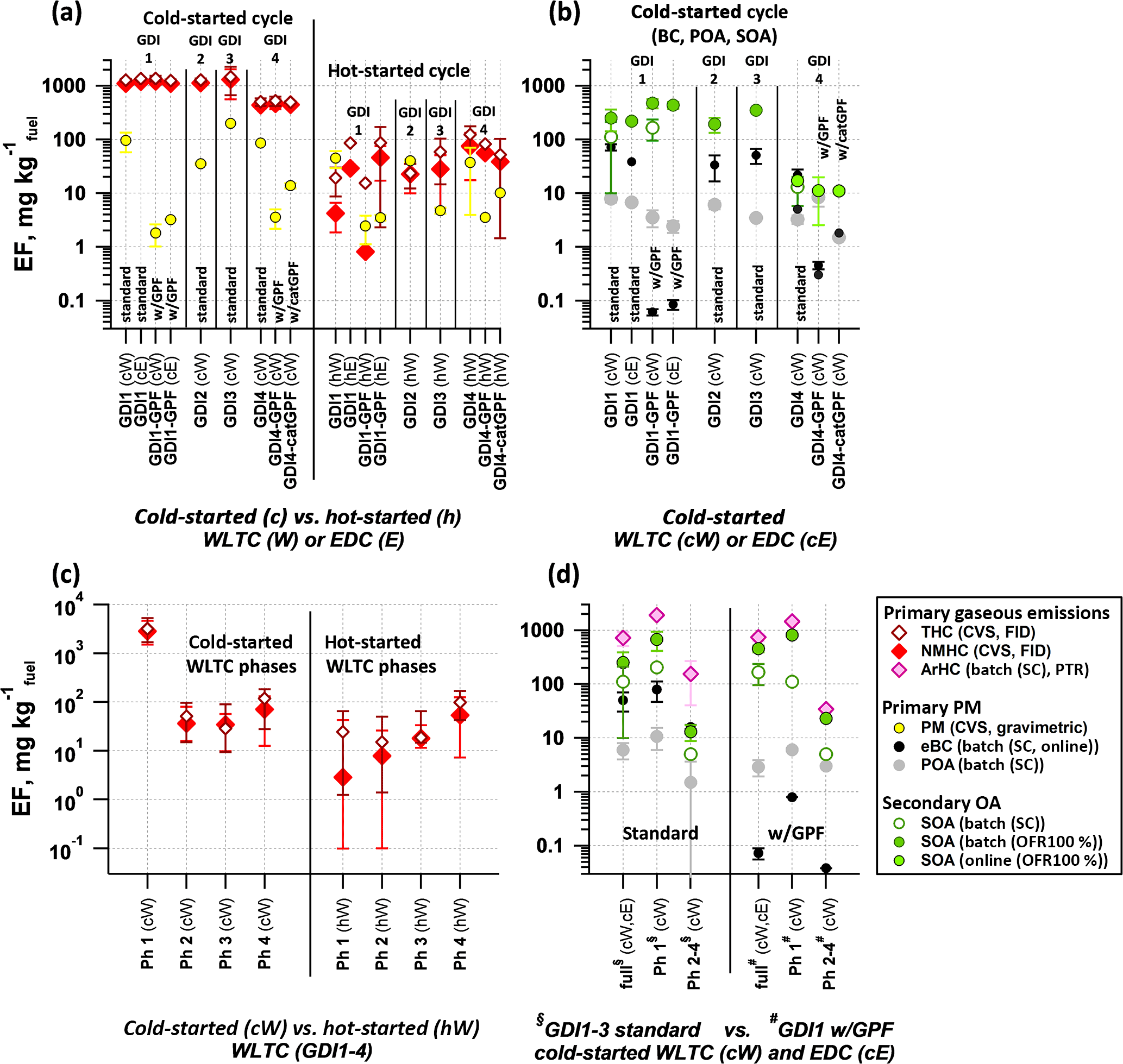
Figure 2Emission factors (EF) of pollutants from cold-started and hot-started test cycles (WLTC and EDC). Individual cW and hW phases are indicated as Ph 1–4. (a) Total and non-methane hydrocarbons (THC, NMHC) and primary gravimetric particulate matter (PM) from CVS measurements over entire test cycles for different vehicle configuration and test conditions (average ± 1 SD). (b) Primary PM (equivalent black carbon (eBC) and primary organic aerosol (POA)) and secondary organic aerosol (SOA) from SC and OFR-from-SC experiments and from online OFR operation at 100 % UV per vehicle configuration for cold-started test cycles (average ± 1 SD). (c) THC and NMHC of cW and hW experiments from (a) separated into individual cycle phases (median and P25–P75 range are shown). (d) POA, eBC, aromatic hydrocarbons (ArHC) and SOA over the full cW and cE, compared to individual phases of cW from SC batch experiments and OFR-from-SC (average ± 1 SD). (a–d) EF calculation is provided in Supplement Sect. S1. The time-resolved SOA profile from online OFR measurements conducted on GDI4 in 2015 (standard and catGPF) is provided in Fig. S14.
3.1.1 NMHC and THC
No drastic test cycle dependencies (WLTC vs. EDC) were observed in terms of NMHC or THC EFs for cold-start conditions (cW vs. cE). The comparison for hot-started cycles (hW vs. hE) was not conclusive, but indicated eventually lower EFs during hE for GDI1. Differences between cold- and hot-started tests were more dramatic: EFs of primary NMHC and THC were reduced by a factor of 90 for GDI1-3 under hot-started conditions compared to cold-started tests (Fig. 2a and c). Median NMHC EFs were 1132 mg kg (cW) and 12.9 mg kg (hW). EFs from cold-started WLTC (cW) for GDI1-3 were clearly dominated by Ph 1 (cW, 4663 mg kg, which exceeded all other test conditions by 2 to 4 orders of magnitude. For GDI4 we found lower total emissions during cold-started cycles compared to other vehicles (∼ factor of 3 lower than GDI1-3, median NMHC EF (cW): 434 mg kg and a smaller difference between cold- and hot-started cycles. For GDI4, the cW NMHC EF was only 8 times higher than from hW, rather than 90 times as for GDI1-3. Instead, when looking at the total NMHC EF of hW, GDI4 exceeded those of GDI1-3 (the median for hW (GDI4) was 55.7 mg kg. This remained true for individual cycle phases. Comparing Ph 1 of cW and hW to Ph 2, 3 or 4 of cW and hW for different vehicle standards revealed that, except for Ph 1 (cW), GDI4 had higher EFs during all other phases than GDI1-3 (factor of 2–30, with the biggest difference found for Ph 2–4 (hW)). The corresponding median data were 4663, 0.1, 23.8 and 1.6 (for GDI1-3, Ph 1 (cW), Ph 1 (hW), Ph 2–4 (cW) and Ph 2–4 (hW) respectively) and 1507, 2.2, 56.8 and 41.1 mg kg (for GDI4). Lower cold-start emissions of GDI4 compared to other vehicles may be explained by differences in the catalytic after-treatment system, the location of the catalyst and/or reduced cold-start enrichment. In terms of NMHC and THC EFs, GDI4 is in line with Euro 6 vehicles, for which regulation also focuses on the reduction of the cold-start hydrocarbon emissions. GPF retrofitting did not affect the NMHC or THC EFs for either GDI1 or 4 under cold-started conditions, as further discussed in Sect. 3.2.
3.1.2 Primary PM (gravimetric PM, eBC, POA)
Primary PM emissions appeared less dramatically affected by the differences between cold- and hot-started cycles compared to above-discussed NMHC and THC EFs (Fig. 2a). Selective sampling of phases of the cold-started cW into the SC (Fig. 2d) and time-resolved measurements (Fig. 3) indicated that significant eBC was emitted during cold-engine start-up (Ph 1 cW). Primary PM emissions were, however, not as strongly reduced during hot-engine conditions (see Ph 2–4 from cold-started cycle as well as hW in Figs. 2a, b, d and 3). The total PM emitted by vehicles in standard configuration was dominated by eBC rather than POA (Fig. 2b) and a POA-to-eBC ratio similar to diesel engines not equipped with DPFs, as also found by Saliba et al. (2017). PM measured in the batch samples (sum of eBC and POA) are compared with gravimetric PM analysis of filters sampled from the CVS in Fig. S16, and chemical analysis of PM samples is further presented in Muñoz et al. (2018). Significant effects in the primary PM EFs were induced by the application of GPFs as discussed in Sect. 3.2.
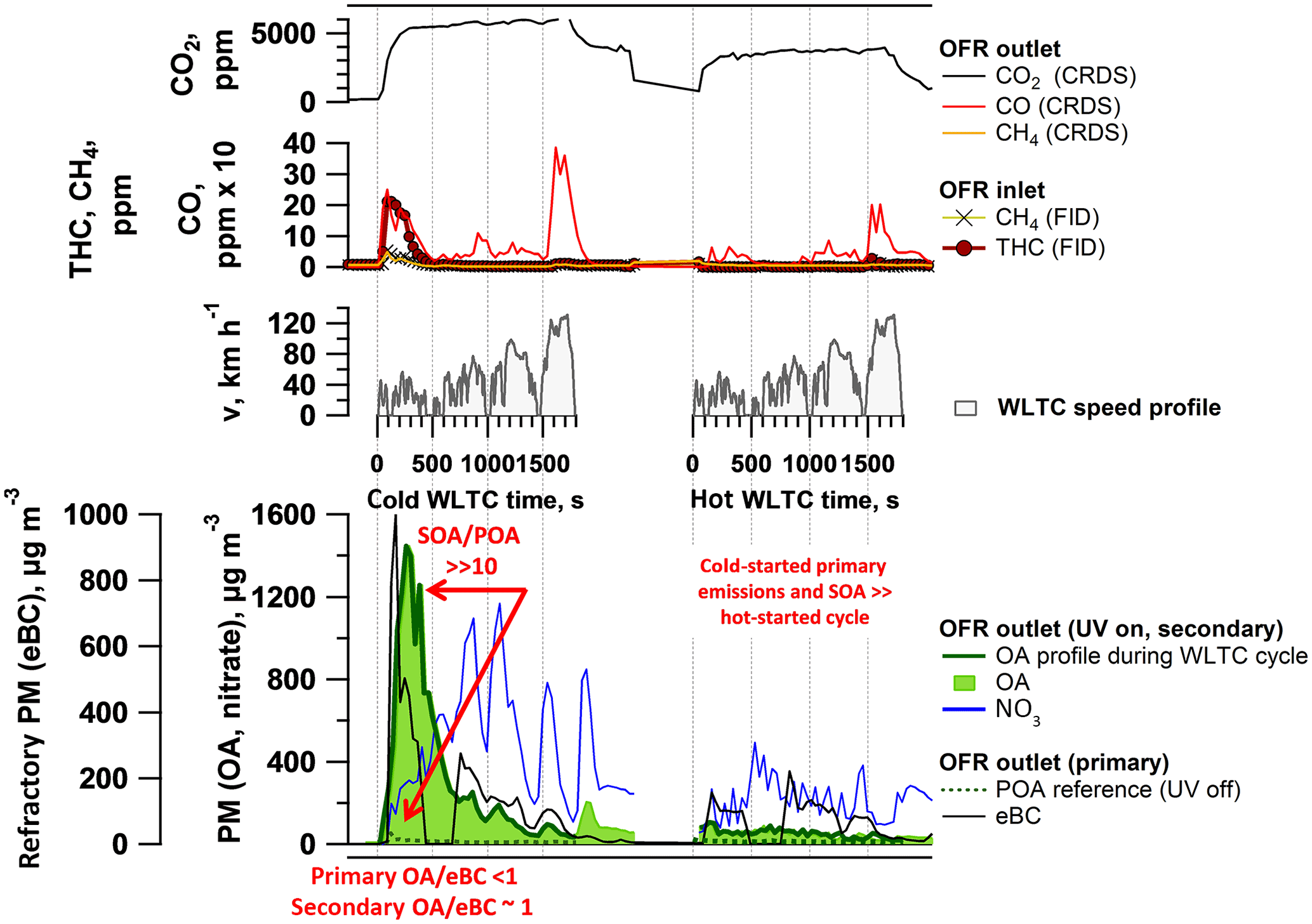
Figure 3Time-resolved aging of cold- and hot-started emissions (WLTC) (GDI1, standard configuration, Expt A2, extended version in Fig. S7). Top: WLTC speed profile. Bottom: OA profile during WLTC presenting the OA measurement during the 30 min driving test with OFR at 100 % UV intensity; due to a delay in the OFR, the signal after the WLTC is finished is displayed as well), nitrate aerosol (inorganic, ammonium nitrate, displayed is only NO3), POA and equivalent black carbon (eBC). Further experiments (A1 a repeat of GDI1 in standard configuration, Fig. S6, and B1 (Fig. S8) and B2 (Fig. S9), which are experiments of GDI1 equipped with GPF) are presented in the Supplement. Time-resolved profiles of GDI4 in standard configuration and with catGPF are provided in Fig. S14.
3.1.3 Secondary organic aerosol
Emissions of all cold-started vehicles, technologies and driving tests showed significant SOA formation upon photochemical oxidation (Fig. 2b), in line with other studies on GDI as well as port-fuel injection systems (Platt et al., 2017; Gordon et al., 2014a; Nordin et al., 2013; Saliba et al., 2017; Zhao et al., 2018). The findings were consistent with the above observation that NMHC and aromatic hydrocarbon EFs (determined by the PTR-ToF-MS; see Fig. 2d) were significantly higher during cold-started cycles compared to other conditions. Hot-engine emissions (Ph 2–4 sampling from cold-started WLTC, as presented in Fig. 2d) also resulted in SOA formation, which was, however, 20–50 times lower in terms of EFs than SOA formed from Ph 1 sampling of a cold-started WLTC. This is likewise in agreement with the trends indicated by the phase-dependent NMHC EFs (Fig. 2c). Also the SOA production factors for GDI4 (median: 12 mg kg were around a factor of 20 lower than the average SOA production of GDI1-3 (Fig. 2b) (median: 222 mg kg. The observed SC SOA (on average 13–170 mg kg was in line with previously aggregated data (e.g., a median 60 and range ∼ 10–400 mg kg, as reported in Jathar et al., 2014) and with our previous findings (range ∼ 6–70 mg kg, Platt et al., 2017). OFR experiments resulted in higher SOA values compared to SC experiments (OFR SOA on average 11–500 mg kg: this was in part due to the higher OH exposure which led to more reacted precursor mass and higher OA loadings. High OA loadings induced partitioning effects (Pankow, 1994; Donahue et al., 2006), which needs to be considered when comparing OFR and SC data. This can be done by comparing SOA data as a function of OA, as presented in Sect. 3.6, Fig. 6. Other differences which may affect the measured SOA mass within the two systems (including vapor losses) are discussed in Sects. 2.2.5 and 3.6.
While engineering measures to reduce cold-start emissions from GDI4 were effective to reduce SOA EFs, and the lower SOA EFs of hot-engine conditions indicated the relevance of a functional after-treatment system to reduce SOA, GPF retrofitting appeared ineffective under cold-started conditions.
3.2 Effect of GPFs on pollutants
Figure 2 provides EFs of GPF-retrofitted vehicles compared to standard configuration, as discussed in Sect. 3.1. We found that gravimetric PM was significantly reduce by the retrofitted GPFs tested on GDI1 and GDI4 (reduction was 98, 96 and 84 % for GDI1-GPF, GDI4-GPF and GDI4-catGPF during cW, respectively; the corresponding hW reduction was 96, 91 and 73 %). The significant primary PM reduction was linked to the removal of the non-volatile eBC fraction (Fig. 2b, d), which dominated the total primary PM and for which reduction values yielded > 99, 94 and 64 % for GDI1-GPF, GDI4-GPF and GDI4-catGPF, respectively, during cW.
Retrofitted GPFs (including catGPF downstream the standard TWC) appeared also to reduce the POA fraction, but the effect was smaller (by 54 to 64 % in three tests, but with a POA enhancement in a fourth test, which we cannot robustly interpret; all data correspond to cold-started cycles). POA removal is more complex, given that POA has a wide range of volatilities and may thus encounter a particle filter in either vapor or particle phase. Only the low-volatility POA fraction may be efficiently removed by filtration, while more volatile material passes through the filter as vapor and condenses when the exhaust is cooled in the ambient air. GPFs did not affect FID-based NMHC (Fig. 2a), aromatic hydrocarbon EFs (Fig. 2d) or the PTR-ToF-MS-based NMOC composition during the cold-started cycles (discussed later in Fig. 4). We have indications for GPF-induced hydrocarbon reduction during hot-engine conditions (by 20–80 % for the FID-based NMHC EFs measured from the CVS system) and believe this deserves further attention in follow-up studies. The retrofitted GPFs did neither reduce the SOA EFs under cold-started conditions (Figs. 2b, d, and S14). SOA reduction requires hence additional after-treatments to remove NMHCs or selected NMOCs, such as reduced cold-start enrichment or engine/catalyst pre-heating. Significantly lowered SOA EFs of GDI4 and during Ph 2–4 SOA experiments are indicated by such engineering measures (Sect. 3.1). No effects of the GPFs were observed on SOA yields or bulk chemical composition of cold-started tests, detailed later (Sects. 3.6–3.7).

Figure 4PTR-ToF-MS derived NMOC composition (cold-started cycles). Data collected by batch sampling (SC) or during online measurements. (a) Mass spectrum of GDI1 emissions (standard configuration) sampled into the SC during a cold-started WLTC (cW). (b) Relative composition of the PTR-ToF-MS derived NMOC fraction (which makes up 65 % ± 15 of the FID-based NMHC signal on a carbon basis for cW, cE, Ph 1(cW)), (c) total ArHC EFs (which make up 49 ± 8 % of the FID-based NMHC signal on a carbon basis for cW, cE, Ph 1(cW)) and (d) relative contribution of the eight dominant ArHC, which correspond to 96.7 ± 3.3 % of the total ArHC signal for cW, cE, Ph 1(cW)). (b–c) Data correspond to vehicle exhaust for GDI1 (experiments A–D), GDI2 (experiment E), GDI3 (experiment F) and GDI4 (experiment G) sampled into the SC during full cW and cE driving tests, or individual phases of cW or measured “online”. The identifier in parenthesis specifies individual SC experiments described in Tables S4–S7. Note that the total NMOC levels for Ph 2–4 (cW) were about 1/10 of full cW and Ph 1 (cW) concentrations only and measurements close to the background (i.e., not significantly different from 3 SD of the background).
3.3 Time-resolved SOA formation in the OFR during dynamic test cycles
CVS and batch sampling of the individual cold-started WLTC phases indicated the highest emission of SOA precursors and SOA formation during cold-started Ph 1 (cW), as detailed in Sect. 3.1.3 and Fig. 2d. This was confirmed by time-resolved SOA profiles from aging of the emissions in the OFR online during the driving cycles, which we show in Fig. 3 for cW and hW tests using GDI1 in standard configuration. The emissions were exposed to OFR photochemistry, with UV intensity at 100 %. Particulate OA and nitrate (denoted NO3) were monitored downstream the OFR; for the cold-started cycle, the POA signal measured during a separate experiment with UV off is shown for reference. The large difference between the OA and POA traces indicated that the observed OA was predominantly SOA. During the cold-started cycle, we found significant SOA formation during Ph 1 (i.e., start and low speed) and to a lesser extent during Ph 2–4 (simulated highway driving), which confirmed our observations from Sect. 3.1.3 and Fig. 2d. The peak at engine start was observed during all cold-start vehicle tests, regardless of vehicle, driving cycle or GPF retrofit, while the small peak at the end of high-speed/extra-urban driving appeared inconsistently. The latter is related to a delay of the OFR signal by the residence time in the reactor, as also observed by Zhao et al. (2018) and might potentially also be caused by a delay of SOA-forming species which are retained on surfaces (Pagonis et al., 2017). The SOA signal correlated with hydrocarbon measurements at the OFR inlet (Figs. S6–S9). The duration of the SOA peak observed at the engine start was likely artificially increased by OFR residence–response timescales and reflects the first few seconds to minutes, prior to catalyst light-off, rather than representing consistently high emissions throughout Ph 1. Supporting this explanation, the hot-started cycle (in which the catalyst operated efficiently from the beginning of the test) did not exhibit any significant emission of NMHC (Fig. 2c) and resulted in relatively little SOA formation when investigated online. Hence, also during online measurements, cold-start emissions appeared to dominate the total GDI SOA burden.
Time-resolved SOA data from 2014 were not used quantitatively herein due to instabilities with the OH exposure throughout the driving cycle, lower OH exposure during high emissions periods and potential impacts by photolysis and competing non-OH processes, as discussed in Supplement Sect. S4, Eq. (S2) and Figs. S11–S12 (Peng et al., 2015, 2016; Li et al., 2015). Further, those data were potentially impacted by an NO influence on the oxidation regime (high vs. low NO levels or NO3 radical formation; discussed in Supplement Sect. S4 and Peng and Jimenez, 2017). This was caused by the low dilution ratio we applied in 2014 (one ejector dilutor, 1 : 8, and additional 1 : 2 at OFR entrance). For the experiments conducted in 2015 such experimental artifacts were reduced by using a higher dilution ratio (two ejector dilutors in series, each 1 : 8 and additional 1 : 2 at OFR entrance). Time-resolved data from 2015 collected with GDI4 were integrated to derived EFs labeled “online, OFR100 %” (Figs. 2b, 4) and agreed well with data derived from corresponding SC experiments. While we cannot rely on an absolute quantitative use of our 2014 data from time-resolved measurements, the relative profile indicating that total SOA was dominated by the cold start-up remains true regardless of those effects and was confirmed in the 2015 data set (Fig. S14). Future work should investigate the quantitative use of online OFR data in further detail for additional quantification of cold- and hot-start contribution of SOA to the total SOA burden; a discussion of the associated technical issues (including also the condensational sink as well as the equilibration time inside the OFR reactor) has been recently published by Zhao et al. (2018).
3.4 Primary NMOC composition investigated by PTR-ToF-MS
3.4.1 Dependence on vehicle test conditions
Figure 4a shows the average NMOC mass spectrum as obtained by the PTR-ToF-MS measurements for exhaust from GDI1 over a cold-started WLTC. The relative composition over all test conditions (driving cycles and phases, vehicle configuration including GPF retrofits) is given in Fig. 4b. Figure 4c summarizes ArHC EFs, and Fig. 4d provides the relative ArHC composition of the most dominant species (a detailed description is provided later). Gasoline as a fuel is mainly composed of aliphatic compounds and ArHC with 7 to 10 carbons (making up roughly 35 % of the fuel volume). The exhaust mass spectral composition from cold-started driving tests appeared instead dominated by surviving fuel additives (ArHC and MTBE), together with incomplete combustion products (ArHC and short-chain aliphatics). The composition was strongly dependent on the driving cycle phase, with ArHC contributing on average 70 % of the total signal of Ph 1 (cW) and the full cycles (cW, cE). Instead, they constituted on average only 14 % in Ph 2–4 (cW). (Note that the NMOC concentrations for Ph 2–4 (cW) were close to our background measurements, i.e., the signal was not significantly different from 3 standard deviations of the background measurement.) ArHC EFs during Ph 1 (cW) were more than 1 order of magnitude higher than Ph 2–4 (cW) EFs. As we showed above, GPF retrofitting did not reduce NMHC or ArHC EFs (Figs. 2, 4c); likewise it had no distinct influence on the overall NMOC composition (Fig. 4b).
3.4.2 Speciation and carbon quantification
In the following we speciate the chemical composition and establish a closure between measurements based on the FID and the PTR-ToF-MS, in order to quantify our potential SOA precursors for Sect. 3.5–3.7. A small number of ArHC ions dominated the mass spectrum and relative composition for full WLTC and Ph 1 (cW), specifically benzene (, integer m∕z 79, denoted BENZ), toluene (, m∕z 93, TOL), o-/m-/p-xylene or ethylbenzene (, m∕z 107, XYL/EBENZ) and C3-benzenes (, m∕z 121, C3BENZ). Their rate constants are shown in Table 2. Relevant additional aromatic peaks corresponded to C4-benzenes (, m∕z 135, C4BENZ), naphthalene (, m∕z 129, NAPH), styrene (, m∕z 105, STY) and methyl-styrene (, m∕z 119, C1STY). While our primary ionization pathway was via H3O+, the ion source produced up to 5 % unwanted , which enabled further pathways (Amador Muñoz et al., 2016; Jordan et al., 2011; Knighton et al., 2009). Signals assigned to pathways were excluded from our analysis (Supplement Sect. S5).
Table 2NMOC information (list of dominant peaks).
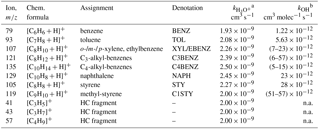
Ions are referred to with their integer mass-to-charge ratio (m∕z) for simplicity, but are identified based on the HR derived exact m∕z instead. n.a. indicates not applicable. a from Cappellin et al. (2012). b kOH from Atkinson and Arey (2003). Range in brackets corresponds to isomers.
The eight above-identified ions comprised 96.7±3.3 % of the total ArHC and 69.5±19.7 % of the total NMOC mass signal in µg m−3 for full cW, cE and Ph 1 (cW); Ph 2–4 (cW) fractions were 65.2±9.8 and 13.9±12.1 %, respectively. Oxygenated ArHC, such as phenolic compounds and benzaldehyde, made up an additional 1.2±2.0 % of the total ArHC fraction for cold-started conditions (cW, cE, Ph 1 (cW)). Their relative contribution increased under hot-engine conditions (Ph 2–4 (cW): 5.9±1.2 %). Also, GDI4 exhibited enhanced contribution of oxygenated ArHC compared to GDI1-3.
The carbon content of the quantified ArHC corresponded to 48.8±7.6 % of the FID-derived NMHC signal assuming equal response factors on the FID, for full cW, cE and Ph 1 (cW). (Note that the ratio of total NMOC mass in µgC determined by the PTR-ToF-MS to NMHC measured by the FID (after subtracting CH4 as measured by the Picarro CRDS) is 0.65±0.15 as average of cW, cE, Ph 1 (cW). The NMHC–NMOC comparison for data for Ph 2–4 is not presented due to interferences on FID measurements of oxygen-containing hydrocarbons.) The high ArHC contribution to the GDI emissions observed here are in line with reports by, for example, Zimmerman et al. (2016a) and Saliba et al. (2017).
Of the non-aromatic peaks in Fig. 4a, the largest signals occurred at integer m∕z 57 ([C4H9]+), followed by 41 ([C3H5]+) and 43 ([C3H7]+), which taken together made up 7.9±4.8 % for the full cycle (cW, cE) and Ph 1 (cW). A larger fraction (13.2±11.9 %) was observed in Ph 2–4 (cW), i.e., hot-engine conditions. These ions are often fragments of larger molecules and hence not straightforward to assign. Thus, they are labeled as structurally unassigned hydrocarbons here. Frequently, [C3H5]+ and [C3H7]+ are considered fragments of oxygenated parent molecules. In our experiments, however, these ions dominantly derived from propene (C3H6), based on ratios between those ions and [C3H6]+ (Supplement Sect. S5, Fig. S15). The fuel contained 5 (2014) to 8 %vol (2015) of MTBE as an anti-knocking agent which, rather than butene, dominated the significant signal at m∕z 57 ([C4H9]+), which is elaborated in Supplement Sect. S5 further. The carbon content of unspecific fragments ([C3H5]+ (m∕z 41), [C3H7]+ (m∕z 43), [C4H9]+ (m∕z 57)) accounted for additional 4.4±3.0 % of the FID NMHC signal (full cW, cE and Ph 1 (cW)).
Based on the literature reports of, for example, Platt et al. (2013) and Schauer et al. (2002), we expect a significant contribution of ethene (C2H4) to the exhaust hydrocarbons. This, however, cannot be quantified by proton transfer reaction (Gueneron et al., 2015) and together with short-chain alkanes contributes in part to the difference between the NMOC and FID-based NMHC signal (ratio of the two measurements: 0.65±0.15). Further possibilities for parents of above-mentioned potential fragments may also contribute to the missing mass closure, e.g., 41, 43 and 57, and further may also derive from alkyl-substituted monocyclic aromatics, alkenes with > C4 or alkanes (> C10, potentially > C6 if cyclic) (Gueneron et al., 2015; Erickson et al., 2014; Buhr et al., 2002). While we detected small intensities at the masses corresponding to (e.g., 71, 85, 99), we did not observe significant signals corresponding to aliphatic fragmentation patterns above m∕z 57. Signals indicating larger cycloalkanes or alkenes (e.g., most abundant fragments at m∕z 69 for substituted cyclohexane) (Gueneron et al., 2015; Erickson et al., 2014) were not abundant, although they were reported by gas-chromatographic MS techniques in other experiments (e.g., Saliba et al., 2017; Zhao et al., 2016). We cannot fully exclude the presence of those compounds due to the limitations of our measurement principle, and they might contribute to the missing 35 % carbon mass. Their potential relevance for SOA is further discussed in Sect. 3.6, and fragmentation is further discussed in Supplement Sect. S5.
We found a small contribution from oxygenated species (such as small acids and carbonyls), while larger oxygenated molecules were not detected except for traces of benzaldehyde () and methyl-benzaldehyde (). Nitrogen was found in very few species, of which the dominant one was acetonitrile (CH3CN). Due to challenges in its quantification without proper calibration of the PTR-ToF-MS, and its unknown source (including potential outgassing from Teflon sampling lines), it was excluded from our analysis. The carbon content of oxygenated compounds would make up only 3.6±3.9 % of the FID signal assuming a response equal to pure HCs for cW, cE and Ph 1 (cW).
3.5 SOA formation in OFR and SC: oxidation conditions and reacted SOA precursors
Figure 5 shows a typical experiment during which collected primary emissions were sampled from the SC through the OFR (OFR-from-SC), exposed to photochemistry at UV light settings of 100, 70 and 50 %, and characterized in dark conditions (Fig. 5a). Thereafter, photochemistry was initiated in the SC (Fig. 5b).
The emissions of vehicle exhaust contained NO, which can influence the chemical pathways during atmospheric processing, given that the dominance of RO2–NO or RO2–RO2 reactions is driven by NO levels. NO-to-NOy ratios are presented in the top panels of Fig. 5. NO was converted rapidly to NO2 (and further to HNO3) in the OFR (Lambe et al., 2017) and OFR aging conditions when sampling from diluted exhaust hence were considered “low NO”. At elevated NO levels such as during online operation of the OFR during our 2014 measurements (discussed in Sects. 3.3 and S4) “high NO” conditions may have been reached as defined by Peng and Jimenez (2017). Based on Platt et al. (2014), RO2 radicals predominantly react with NO when the concentration of NO is higher than only 1 ppb in the SC. Before starting SC aging by injecting HONO and initiating photochemistry, we titrated NO present in the SC to NO2 using O3. NO levels in the SC typically dropped to the detection limit (< 1 ppb) within few minutes of photochemistry. The total NOy signal increased with time of SC experiment, which we relate to the formation of HNO3 from primary NOx and continuous injection of nitrous acid (HONO). The presence of NO2 could not be unambiguously quantified. We classified our SC experiments as “low NO” conditions, although initial NO concentrations might be higher than in the corresponding OFR experiments.
Due to photochemistry, reactive NMOCs decayed due to reactions with OH radicals (Fig. 5a, b, middle panel), while OA and secondary nitrate mass increased in turn (bottom panel). While in terms of abundance of potentially SOA-forming precursors toluene (TOL) and xylenes and ethylbenzene (XYL/EBENZ) dominate over benzene (BENZ) and the C3-benzenes (C3BENZ), their OH reaction rates (Table 2) have the opposite trend (C3BENZ > XYL/EBENZ > TOL > BENZ). The reacted ArHC mass at a given OH exposure was governed by the combination of their abundance and their reaction kinetics. At the final OH exposure of (1.4–5.8) × 1011 molec cm−3 s the reacted ArHC mass was dominated by XYL/EBENZ (41±3 %), which together with TOL (33±4 %) comprised more than 70 % of the total reacted ArHC. C3BENZ (13±2 %) and BENZ (7±3 %) provided smaller contributions, and C4BENZ, STY, C1STY and NAPH accounted for additional 5 %; other compounds were not considered (fractions are provided in Fig. S5; OH exposure data at the end point of SC experiments and for the OFR are provided in the caption of Fig. 6 and in Fig. 7).
NO3 to OA ratios as a surrogate to describe NH4NO3 formation were 4.00±2.11 in the SC and comparatively lower in the OFR (0.43±0.26).
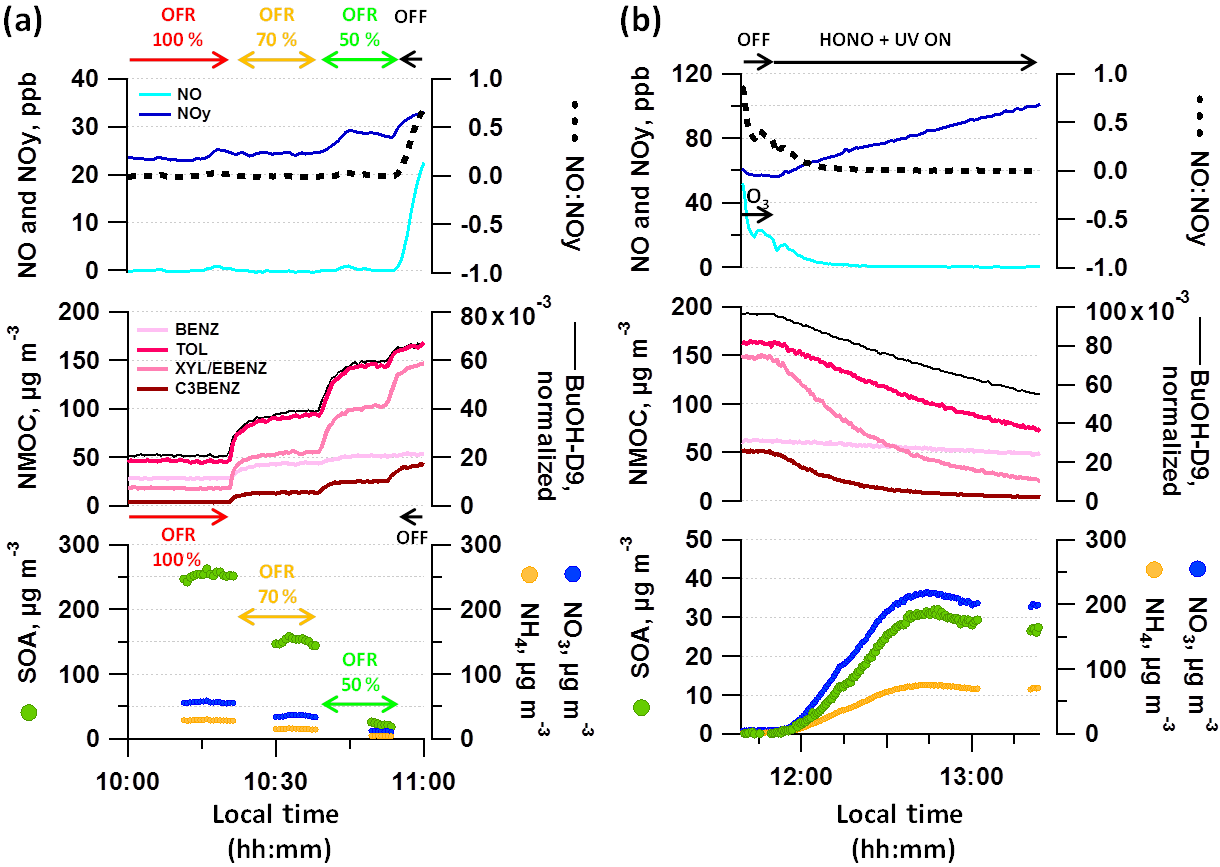
Figure 5Typical OFR-from-SC and SC photochemistry experiment. Decay of dominant SOA precursors (benzene (BENZ), toluene (TOL), o-, m- and p-xylene (XYL) or ethylbenzene (EBENZ), C3-benzenes (C3BENZ)) through photochemistry and associated SOA formation in (a) OFR (sampling from SC batch at different UV intensities, displayed is experiment D3) and (b) SC (displayed is experiment A2). (a–b) UV status, O3 and HONO injection are indicated along with the NO : NOy ratio and the OH tracer BuOH-D9. Reacted ArHC fractions are provided in Fig. S5 per experiment. Local time is given in intervals of 15 min.
3.6 Effective SOA yields
Effective SOA yields (Ye) as a function of absorptive mass (Pankow, 1994; Donahue et al., 2006) are displayed in Fig. 6. For the GDI exhaust, our Ye assumed BENZ, TOL, XYL/EBENZ, C3BENZ, C4BENZ, NAPH, STY and C1STY as sole SOA precursors and we focused on tests from cold-started GDI1-3 (i.e., for full cW, cE; and Ph 1 (cW)), while GDI4 or hot-engine conditions, i.e., Ph 2–4 (cW), were not included in our analysis. This is because the concentration levels were close to our background measurements. However, we would like to highlight that Zhao et al. (2018) recently reported higher effective SOA yields for hot-engine conditions compared to cold-engine conditions, which reflects also our observations. All yields (for exhaust as well as separate precursors) increased as a function of the suspended OA, reaching 0.8–1 for OFR vehicle exhaust experiments with OA loadings above 300 µg m−3. In the atmospherically more relevant range of 10 to 100 µg m−3, yields spread from a few (< 15 %) to 20–50 %. Detailed discussions are provided later. In brief, we found the following:
-
SC- and OFR-derived effective yield curves for GDI exhaust agreed within our experimental variability and had a trend for higher yields in the OFR than the SC (or, vice versa, lower yields for the SC than the OFR) (Fig. 6c, Sect. 3.6.1). Neither a distinct difference between Ph 1 (cW) SOA and the full cycle (cW, cE) SOA nor an explicit effect of GPF retrofitting was observed.
-
GDI vehicle exhaust effective SOA yields (SC and OFR) appeared relatively higher than our reference measurements with specific SOA precursors, by up to a factor of 2, with larger discrepancies for the OFR and smaller discrepancies for the SC. This is detailed further below (Fig. 6a, Sect. 3.6.2).
-
OFR SOA yields of toluene, o-xylene and 1, 2, 4-TMB and their mixtures were in good agreement with those of other OFR studies (m-xylene; Ahlberg et al., 2017) and SC studies (benzene, toluene, o-xylene; Li et al., 2016a, b, Fig. 6b).
3.6.1 SC vs. OFR yields of GDI exhaust (Fig. 6c)
Aging of GDI vehicle exhaust in the SC and the OFR resulted, within our experimental variability, in similar effective SOA yield curves. They exhibited a trend towards higher values for OFR than SC experiments (or, vice versa, lower values for the SC) (Fig. 6c). Yields determined in the SC experiments were, however, variable among themselves and investigations of agreement between SC- and OFR-derived yields consequently were a function of the chosen reference point. We believe that experiments A2, A3 and B3 (as labeled in Fig. 6 and Table S4) are reliable data points for comparison, while experiments A1, B1 and B2 are potentially associated with higher uncertainties (further discussed in the paragraph following the next).
Yields are expected to be underestimated by factors of 1.5–2 (SC) and 1.25 (OFR) (Platt et al., 2017; Palm et al., 2016) due to influences of vapor wall losses. Taking those correction factors into account reduces the discrepancy between the two systems. The relative contributions of species to the reacted ArHC fraction were not significantly different between the systems (Sect. 3.5, Fig. S5). On average they agreed by a factor of 1.0±0.3. However, other plausible explanations exist for the remaining gap in the yields. While higher initial levels of NO in the SC experiments might suppress SC SOA formation, as recently discussed by Zhao et al. (2017), the more likely scenario in our experiments is that the higher OH concentrations in the OFR (107 molec cm−3 in the SC, vs. 108 to 109 in the OFR) led to multiple OH interactions (Molteni et al., 2018) and thereby enhanced the OFR yields. This is also supported by the higher H : C found in OFR SOA (see discussion in Sect. 3.7).
As mentioned earlier, we also investigated o the variability among SC yields, which indicated a correlation of higher SOA yields with higher initial SC NO levels, such as for experiments A1, B1 and B2 (Fig. 6, Table S4). This is contradictory to common knowledge and recent work by Zhao et al. (2017). The higher initial NO levels, however, also correlated with higher concentrations of secondary NH4NO3 in the SC (Table S4, using the NO3 as a surrogate). Presenting SC yields as a function of in Fig. S13 appeared to decrease variability among SC yields, indicating NH4NO3 dependencies for those three experiments (A1, B1, B2). Given that the high NH4NO3 concentration in these experiments was outside our -AMS interference calibration, data may be associated with a positive mass bias even after correction (Pieber et al., 2016). Neglecting experimental artifacts would allow for a speculation on the contribution of inorganic nitrate and the associated water as absorptive mass (Stirnweis et al., 2017) or the (unwanted) influence of NO3 radicals at relatively higher concentrations of initial NO (Schwantes et al., 2017). A detailed analysis is, however, beyond the scope of our study.
3.6.2 GDI exhaust SOA yields in comparison to specific precursors (Fig. 6a)
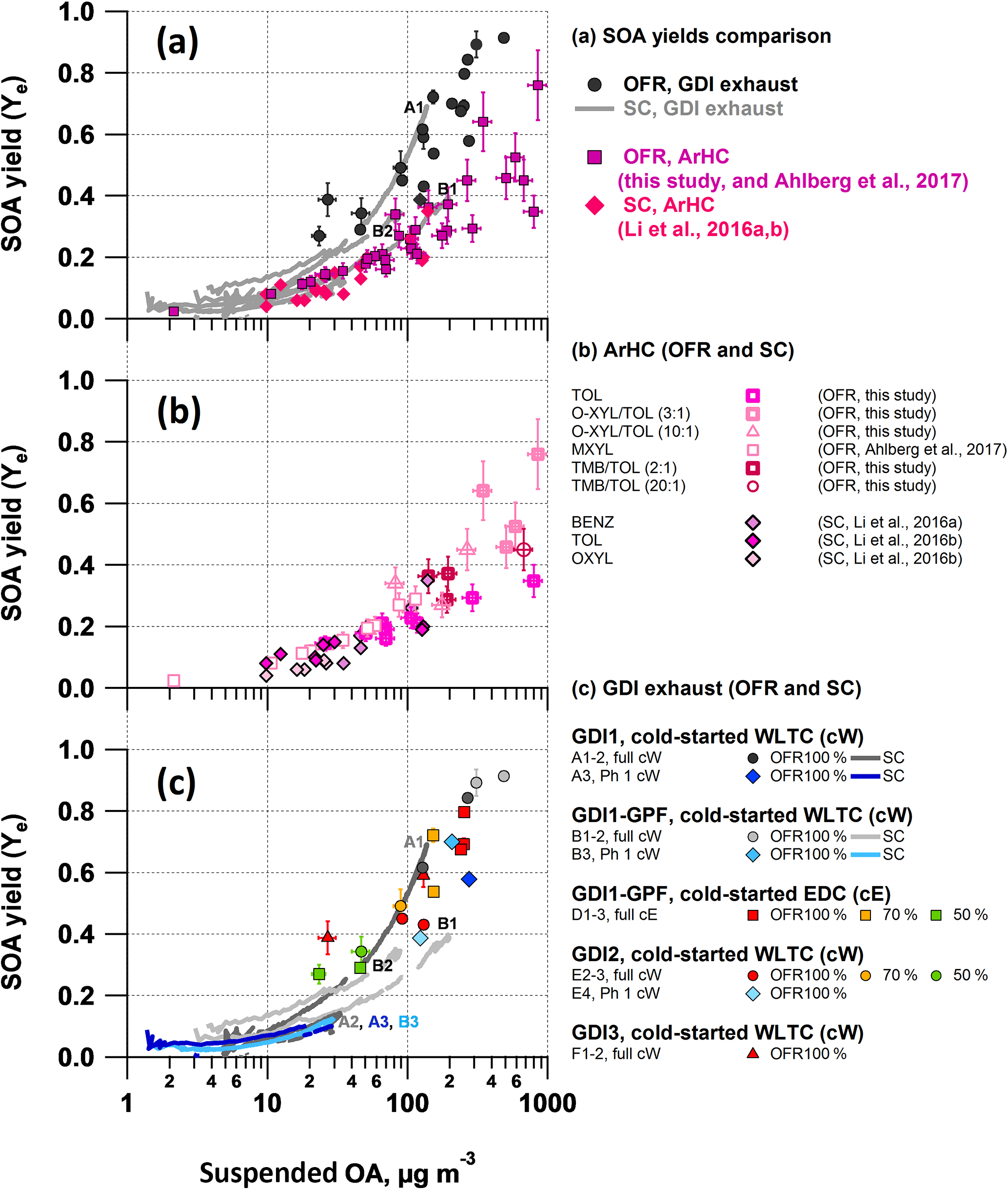
Figure 6Effective SOA yields. Vehicle exhaust from GDI1-3 (full cW, full cE, Ph1 (cW)) photochemically aged in the SC and OFR-from-SC compared to effective SOA yields from selected ArHC (toluene, o-xylene, 1,2,4-TMB) photochemically aged in our OFR (this study, without NO; m-xylene data from Ahlberg et al., 2017) and in a SC (benzene, toluene, o-xylene from Li et al., 2016a, b, without NO). (a) all data combined; (b) OFR (data from this study are also provided in Table S8) and SC yields of single ArHC or mixtures; (c) vehicle exhaust photochemically aged in SC and OFR-from-SC (average ± 1 SD of AMS OA measurement during stable conditions). Error bars on data from OFR represent the variability of the measurement. SC yield curves per experiment are presented in Fig. S13 and potential factors enhancing yields in experiments A1, B1 and B2 (Table S4) are discussed in Sect. 3.6.1. (a–c) OH data are given in Fig. 7 and summarized here: OH exposures up to 1.4 × 1011 molec cm−3 s, after ∼ 2 h of SC photochemistry (average [OH] = 2 × 107 molec cm−3). OFR100 %: [OH] = (2.7–5.2) × 109 molec cm−3; [OH]exp= (3.0–5.8) × 1011 molec cm−3 s (at ∼ 8 ppm O3). OFR70 %: [OH] = (1.4–2.2) × 109 molec cm−3; [OH]exp= (1.6–2.5) × 1011 molec cm−3 s (at ∼ 3 ppm O3). OFR50 %: [OH] = (0.28–0.44) × 109 molec cm−3; [OH]exp= (0.31–0.49) × 1011 molec cm−3 s (at ∼ 0.7 ppm O3). The maximum OH exposure in the SC corresponds to the range of green to orange colored OFR data points in panel (c); see Fig. 7.
GDI vehicle exhaust effective SOA yields from SC and OFR appeared higher than our reference measurements with specific SOA precursors, again from SC and OFR. Effective yields of vehicle exhausts were in the range of those from single precursors, particularly when considering SC experiment, but with a higher discrepancy for the OFR experiments (Fig. 6a). To explain the remaining discrepancy, which was up to a factor of 2, we focus on the following two hypotheses:
-
Our analysis does not sufficiently account for all precursors (see also Sect. 3.4). Our calculated effective SOA yields assume that all relevant SOA precursors were identified and their decay quantified, as defined in Eq. (2). We were able to explain 65%±15% of the total non-methane hydrocarbon signal with the carbon found in the PTR-ToF-MS measured NMOCs and used the aromatic fraction (49±8 %) as SOA precursors. This approach covers a significant fraction of likely SOA precursors. While both the aromatic (Odum et al., 1997; Ng et al., 2007b; Hildebrandt et al., 2009; Loza et al., 2012; Platt et al., 2014) and the aliphatic (especially alkanes) (Lim and Ziemann, 2005; Loza et al., 2014) species are known exhaust constituents and may form SOA, aliphatic species are relevant only if their carbon chain is sufficiently long and does not substantially fragment during reaction. Short-chain alkanes (< C8) exhibit only low SOA yields at typical ambient OA levels (Jordan et al., 2008). ArHC, starting from the simplest with C6, instead produce highly oxygenated multifunctional organics with only few OH attacks (Molteni et al., 2018; Schwantes et al., 2017) and are therefore efficient SOA precursors exhibiting high yields. Oxygenated ArHC (phenolic, benzaldehyde) did not appear significant enough (< 1 % of the total NMOCs) to induce yield enhancements and were neglected in our analysis. Further relevant compounds were not included as relevant SOA precursors, although, on average, up to 35 % additional carbon was available in undetected molecules (assuming the PTR-ToF-MS to FID comparison is a valid approach). Parts of those 35 % are certainly not significant for SOA formation, such as ethene and other above-mentioned short-chain aliphatic compounds. While those might contribute significantly to the unidentified carbon fraction, they do not contribute significant SOA mass. Other undetected molecules instead might also form SOA, and leaving them unaccounted artificially increases our calculated effective SOA yields.
Prominent candidates are alkyl-substituted monocyclic aromatic and long-chain aliphatic compounds, as elaborated on in the following. Identified ArHC as determined by the PTR-ToF-MS were classified as VOCs based on their saturation mass concentration (C*) at or above 106 µg m−3 (VOCs) with a small contribution from aromatics (such as naphthalene) in the intermediate volatility organic compounds (IVOC) range (C* = 102−106 µg m−3) (Pandis et al., 2013). While the larger contribution of VOCs than IVOCs to gasoline vehicle exhaust SOA is consistent with Zhao et al. (2016), it also suggests additional substituted monocyclic aromatic IVOCs, which we did not identify. Likewise Nordin et al. (2013) postulated alkyl-substituted monocyclic aromatics previously as relevant precursors. Given that they fragment in the PTR-ToF-MS predominantly by losing the aromatic ring, those compounds could indeed be significant contributors to the 35 % missing carbon mass and would also contribute to SOA. Long-chain aliphatic compounds are likewise plausible, although we found no significant indication in our mass spectra. Further investigations of those species using the PTR-ToF-MS could be performed by inducing other ionization pathways such as by use of as the primary ion source (e.g., Amador Muñoz et al., 2016).
Finally, MTBE was present in significant amounts in the exhaust. It has currently not been considered as a significant SOA precursor, owing to its small carbon number and high volatility. We believe it should be investigated in future work considering it may contribute to SOA when contained in a complex mixture.
-
Reference SOA yields do not accurately represent the complex exhaust emissions. Complex mixtures of hydrocarbons and matrix effects might exhibit SOA yields which differ from single molecules or relatively simple mixtures. The influence of NO on SOA yields has been previously addressed in the literature for biogenic and anthropogenic sources (e.g., Ng et al., 2007a, b) and generally indicates that at higher NO conditions, lower SOA yields are observed. Zhao et al. (2017) confirmed this for gasoline exhaust, and we choose NO-free conditions as comparison points for our yields based on the discussion in Sect. 3.5. Choosing a high-NO reference would enhance discrepancies. Instead, the influence of other exhaust constituents which are absent in our reference measurements, such as the formed NH4NO3, the presence of NO2 and chemical processing by unwanted formation of NO3 radicals (Schwantes et al., 2017), is insufficiently addressed in the literature for a final conclusion. Matrix processes, along with potential nonlinear effects of SOA formation from mixed precursors, should be addressed in future studies. Further, aromatic isomers show a distribution of yields based on carbon number, number of aromatic rings and degree and location of substitution, which are not fully covered by the reference compounds selected for testing. Isomers present in the exhaust may enhance the effective SOA yield relative to the reference measurements. Lastly, benzene contributed less than 10 % to the reacted NMOCs (Sect. 3.5 and Fig. S5) and was therefore not tested separately in our OFR. However, its SOA yield has been reported to exceed that of alkylated analogous compounds, such as xylenes or higher alkylated benzenes (Li et al., 2017; Bruns et al., 2016). Benzene may hence contribute to the enhanced effective SOA yield relative to the reference measurements.
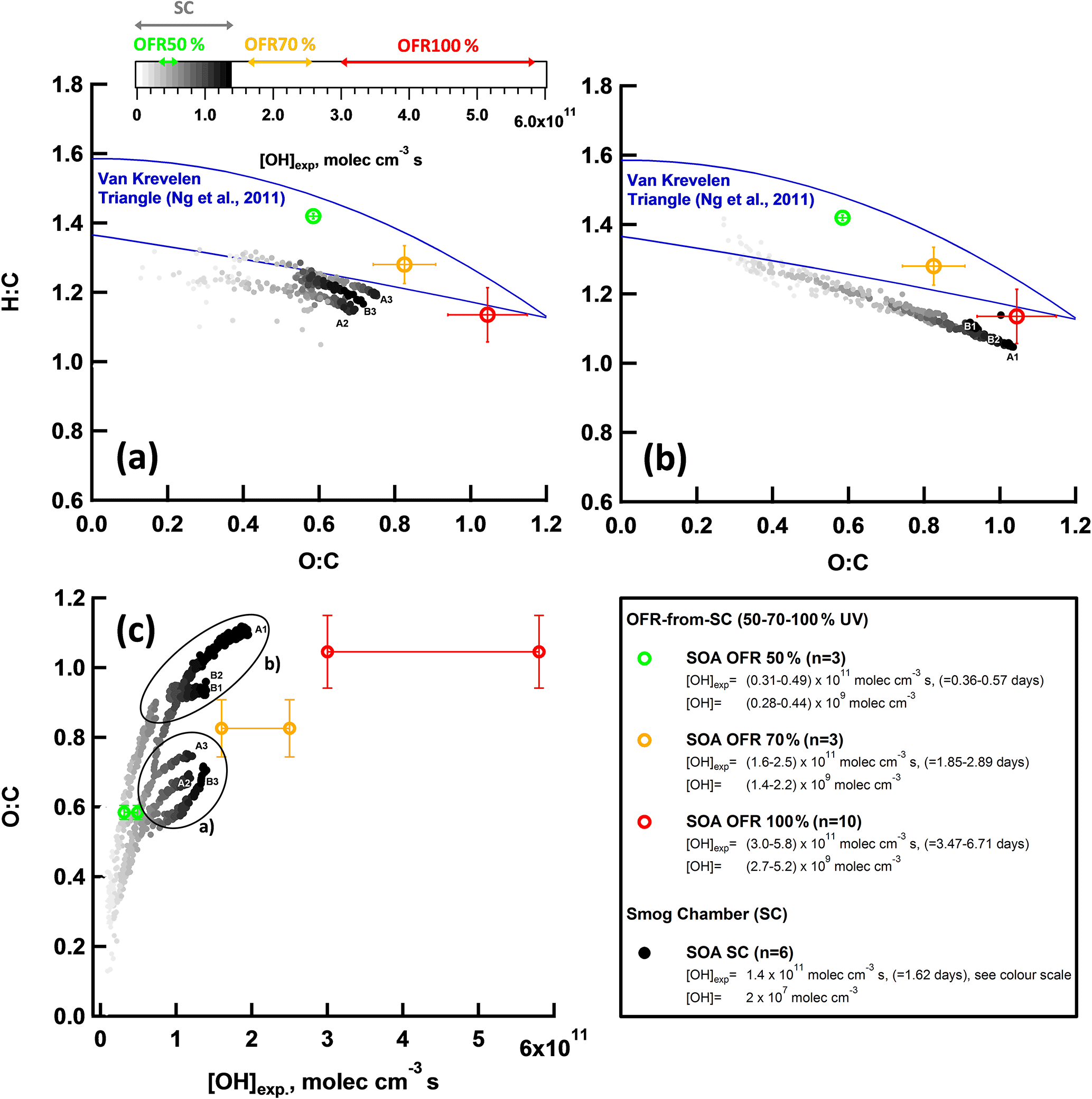
Figure 7Bulk OA composition of SC and OFR SOA. (a–b) Van Krevelen plot (O : C vs. H : C) for SOA formed during SC experiments (n=6, GDI1 standard and w/GPF, cW and Ph 1 (cW)) and OFR-from-SC data points (n=10, GDI1 standard and w/GPF, full cW, full cE, Ph 1 (cW)) at different OFR UV settings (100, 70, 50 %). (a) SC experiment (A2, A3, B3; Table S4) and (b) SC experiment (A1, B1, B2; Table S4), experiments with NH4NO3 levels outside our -AMS interference calibration range (Pieber et al., 2016). The POA contribution was subtracted from the total OA bulk composition; SOA ∕ POA ratios were > 10. The Aiken parameterization (Aiken et al., 2007, 2008) has been applied to HR fitted data. Lines indicate the Van Krevelen space typical for ambient AMS measurements (Ng et al., 2011). Error bars represent 1 standard deviation of measurement variability. (c) O : C of (a) and (b) as a function of [OH] exposure. [OH]exp in days refers to an assumed average ambient [OH] of 106 molec cm−3.
3.7 SOA elemental composition (SC and OFR)
The bulk OA elemental oxygen-to-carbon and hydrogen-to-carbon ratios (O : C and H : C) for GDI exhaust SOA formed in SC and OFR-from-SC experiments at varied OH exposure are shown in Fig. 7. The SOA composition shifted towards higher O : C and lower H : C as a function of OH exposure in both systems. While we found agreement for the O : C between SC and OFR-from-SC at similar OH exposure for three SC experiments (labeled A2, A3, B3 in Fig. 7a, c), the other three experiments exhibited relatively higher O : C at equivalent OH exposure (labeled A1, B1, B2 in Fig. 7b, c). The latter also had higher SOA yields (Sect. 3.6.1) and were characterized by higher NH4NO3 concentrations, which were outside our -AMS interference calibration. We believe data may be associated with a positive bias towards higher O : C even after correction (Pieber et al., 2016). Hence we focused on A2, A3 and B3 and consider our O : C data in general agreement between OFR SOA and SC SOA, when represented as a function of OH exposure. This agreement did not apply for the H : C, however, for which the OFR yielded higher values than the SC. Oxidation products with two more H atoms than the precursor are formed when the aromatic-OH adduct adds an oxygen molecule and the peroxy radical then terminates by a reaction with HO2 or RO2. If the oxidation product contains a C = C double bond, this reaction sequence can be repeated, leaving a second generation oxidation product with four additional H atoms. The formation of highly oxygenated low-volatility products with two and four additional H atoms under high OH concentrations has been shown by Molteni et al. (2018). The higher NO levels in the SC and the higher peroxy radical concentration in the OFR are critical to which termination pathways of the peroxy radical occur. For example, an enhanced reaction termination with HO2 rather than RO2 would increase the H : C in the OFR relative to the SC. Further investigation of those aspects requires information on a molecular level and should be the focus of future comparison studies. GPF retrofitting did not distinctly affect SOA bulk elemental composition, which is in line with no clear effects on NMOC composition, SOA EFs or SOA yields.
We studied exhaust from Euro 4 and Euro 5 GDI vehicles as a function of driving cycles, individual phases thereof and engine temperature (cold-started, hot-started), and evaluated the effect of retrofitted GPFs on primary emissions and SOA. We presented a detailed analysis of primary NMOC composition from PTR-ToF-MS measurements, identified relevant SOA precursors and assessed SC and OFR experiments. Here, we summarize the major conclusions.
For all GDI vehicles, the dominant fraction of hydrocarbon emissions was released during cold-started vehicle tests, before after-treatment systems are at operational temperature. No drastic test cycle dependencies between WLTC and EDC were observed from our tests during cold-started cycles. Instead, EFs of primary NMHC and THC were reduced by up to a factor of 90 under hot-started conditions compared to cold starts, and total emissions were dominated by the pollution during the first few minutes of the driving cycle. Chemically, the emissions of cold-started vehicles were dominated by aromatic hydrocarbons, especially by toluene, xylenes and ethylbenzene, C3-benzenes and benzene. SOA formation was likewise governed by the cold-start emissions, and SOA formation under hot-engine conditions was 20–50 times lower than under cold-engine conditions. These results were independent of the testing protocol, demonstrating that vehicle engineering and the performance of after-treatment systems rather than the driving behavior governed these emissions. Overall, the SOA potential (in terms of an emission factor) agreed with recent literature reports from both GDI and port-fuel injection systems. It appeared that GDI4, which was in line with Euro 6 regulations regarding its NMHC emissions, had a reduced overall and cold-start NMHC EF, but instead its emissions during hot-engine conditions contributed a bigger relative fraction to the total. Additionally, by trend, oxygenated ArHC had a slightly enhanced fraction in GDI4 compared to GDI1-3 exhaust. SOA formation of GDI4 was lower compared to GDI1-3, in line with NMHC reduction induced by reduced cold-start enrichment or improved catalytic after-treatment system. Considering that GDI4 NMHC EFs follow those of Euro 6 vehicles, the determined SOA EFs may be representative of a newer generation of vehicles.
GPF retrofitting efficiently removed eBC, which was the dominant component of primary PM. It also showed effects on the minor POA fraction, which was, however, not as significantly reduced as the refractory PM. Instead, GPF retrofitting did not alter NMHC EF, the chemical gas-phase composition, and it did not reduce SOA formation in our cold-started tests. This result holds likely generally true when GPFs are catalytically inactive and, at cold-started driving cycles, for catalytically active GPFs as well (i.e., when emissions pass through the TWC and the catGPF before light-off temperatures are reached). It implies that while retrofitting GDI vehicles with GPFs will likely result in an important reduction of the total primary PM emissions through removal of refractory material, it will (under conditions similar to our experiments) only to a small extent reduce hydrocarbon emissions, including ArHC, and thereby not directly lead to SOA reduction. Future work should assess GPF and catGPF effects under hot-engine conditions in more detail. Likewise, tests on so-called “four-way catalysts”, i.e., a TWC–GPF combination installed at the location of the current TWC for simultaneous filtration of particulates and catalytic conversion of gaseous pollutants, will be beneficial to understand whether reductions of SOA precursors, SOA and semi-volatile primary PM can be achieved with further optimized systems.
Effective SOA yields from GDI exhaust, while in general agreement considering our experimental variability, tended to be higher for the OFR than the SC (or, vice versa, lower in the SC than the OFR) and were not explicitly influenced by GPF retrofitting. Trends in the elemental O : C of the bulk SOA were related to different OH exposure levels in the two systems. Trends in the H : C indicated instead differences in OFR and SC processing, which call for further investigation on a molecular level. SOA formation from GDI vehicle exhaust appeared dominated by a few ArHC and was not affected by GPF retrofitting. While a significant fraction of the SOA could be attributed to the identified precursors, divergences in the effective SOA yields remained up to a factor of 2 when comparing to specific precursors. This may have diverse reasons including unaccounted precursors (which cannot be detected by PTR-ToF-MS measurements) and complex matrix effects which deserve further attention in follow-up studies.
Data related to this article are available online at https://doi.org/10.5281/zenodo.1305770. This includes emission factors reported in Figs. 2 and 4, SOA yields from Fig. 6, and bulk elemental composition of SOA from Fig. 7. Additional data are available in the Supplement to this article. Further information will be provided upon request to the corresponding authors.
| AMS | Aerosol mass spectrometer |
| ArHC | Aromatic hydrocarbons (including functionalized aromatic hydrocarbons) |
| catGPF | Catalytically active gasoline particle filter |
| cE | Cold-started EDC vehicle test |
| cW | Cold-started WLTC vehicle test |
| eBC | Equivalent black carbon, as determined by Aethalometer measurements |
| EDC | European Driving Cycle (previously known as the “New European Driving Cycle”) |
| FID | Flame ionization detector |
| GDI | Gasoline direct injection vehicle |
| GPF | Gasoline particle filter |
| hE | Hot-started EDC vehicle test |
| hW | Hot-started WLTC vehicle test |
| IVOC | Intermediate-volatility organic compound |
| molec | Molecule |
| NMHC | Non-methane hydrocarbons, i.e., gaseous organic compounds (hydrocarbons) as measured by FID |
| NMOC | Non-methane organic compounds, i.e., gaseous organic compounds as measured by PTR-ToF-MS |
| OFR | Oxidation flow reactor |
| OFR-from-SC | Also referred to as “batch OFR” – OFR continuously sampling from |
| a batch sample previously collected in the SC | |
| Online OFR | OFR deployed online during a driving cycle, connected directly to diluted exhaust |
| PCFE | Particle count filtration efficiency |
| Ph 1 | First phase of WLTC; Ph 1 (cW) refers to first phase of cold-started WLTC |
| Ph 2–4 | Second to fourth phase of WLTC; Ph 2–4 (cW) refers to the second to fourth phase of cold-started WLTC, |
| which are quasi-hot-engine conditions | |
| POA | Primary organic aerosol |
| PTR-ToF-MS | Proton transfer reaction time-of-flight mass spectrometer |
| SC | Smog chamber |
| SOA | Secondary organic aerosol |
| VOC | Volatile organic compound |
| WLTC | Worldwide light-duty test cycle |
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-9929-2018-supplement.
The authors declare that they have no conflict of interest.
This work was funded by the CCEM project GasOMeP
(http://www.ccem.ch/gasomep, last access: 6 June 2018). Many thanks to
our collaborators within the GasOMeP project for their input during
scientific meetings. We also acknowledge support by the Swiss National
Science Foundation (SNF Project 140590 and SNF starting grant
BSSGI0_155846). We would like to thank René Richter for his invaluable
technical support as well as the staff of the Laboratories for IC-Engines and
Exhaust Emission Control of the Berne University of Applied Sciences in Biel,
who conducted the vehicle testing.
Edited by:
Jacqui Hamilton
Reviewed by: two anonymous referees
Ahlberg, E., Falk, J., Eriksson, A., Holst, T., Brune, W. H., Kristensson, A., Roldin, P., and Svenningsson, B.: Secondary organic aerosol from VOC mixtures in an oxidation flow reactor, Atmos. Environ., 161, 210–220, https://doi.org/10.1016/j.atmosenv.2017.05.005, 2017.
Aiken, A. C., DeCarlo, P. F., and Jimenez, J. L.: Elemental analysis of organic species with electron ionization high-resolution mass spectrometry, Anal. Chem., 79, 8350–8358, https://doi.org/10.1021/ac071150w, 2007.
Aiken, A. C., Decarlo, P. F., Kroll, J. H., Worsnop, D. R., Huffman, J. A., Docherty, K. S., Ulbrich, I. M., Mohr, C., Kimmel, J. R., Sueper, D., Sun, Y., Zhang, Q., Trimborn, A., Northway, M., Ziemann, P. J., Canagaratna, M. R., Onasch, T. B., Alfarra, M. R., Prévôt, A. S. H., Dommen, J., Duplissy, J., Metzger, A., Baltensperger, U., and Jimenez, J. L.: O ∕ C and OM ∕ OC ratios of primary, secondary, and ambient organic aerosols with high-resolution time-of-flight aerosol mass spectrometry, Environ. Sci. Technol., 42, 4478–4485, https://doi.org/10.1021/es703009q, 2008.
Amador-Muñoz, O., Misztal, P. K., Weber, R., Worton, D. R., Zhang, H., Drozd, G., and Goldstein, A. H.: Sensitive detection of n-alkanes using a mixed ionization mode proton-transfer-reaction mass spectrometer, Atmos. Meas. Tech., 9, 5315–5329, https://doi.org/10.5194/amt-9-5315-2016, 2016.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Bahreini, R., Middlebrook, A. M., de Gouw, J. A., Warneke, C., Trainer, M., Brock, C. A., Stark, H., Brown, S. S., Dube, W. P., Gilman, J. B., Hall, K., Holloway, J. S., Kuster, W. C., Perring, A. E., Prévôt, A. S. H., Schwarz, J. P., Spackman, J. R., Szidat, S., Wagner, N. L., Weber, R. J., Zotter, P., and Parrish, D. D.: Gasoline emissions dominate over diesel in formation of secondary organic aerosol mass, Geophys. Res. Lett., 39, L06805, https://doi.org/10.1029/2011gl050718, 2012.
Barmet, P., Dommen, J., DeCarlo, P. F., Tritscher, T., Praplan, A. P., Platt, S. M., Prévôt, A. S. H., Donahue, N. M., and Baltensperger, U.: OH clock determination by proton transfer reaction mass spectrometry at an environmental chamber, Atmos. Meas. Tech., 5, 647–656, https://doi.org/10.5194/amt-5-647-2012, 2012.
Barrett, S. R. H., Speth, R. L., Eastham, S. D., Dedoussi, I. C., Ashok, A., Malina, R., and Keith, D. W.: Impact of the Volkswagen emissions control defeat device on US public health, Environ. Res. Lett., 10, 114005, https://doi.org/10.1088/1748-9326/10/11/114005, 2015.
Bond, T. C., Streets, D. G., Yarber, K. F., Nelson, S. M., Woo, J. H., and Klimont, Z.: A technology-based global inventory of black and organic carbon emissions from combustion, J. Geophys. Res., 109, D14203, https://doi.org/10.1029/2003JD003697, 2004.
Borbon, A., Gilman, J. B., Kuster, W. C., Grand, N., Chevaillier, S., Colomb, A., Dolgorouky, C., Gros, V., Lopez, M., Sarda-Esteve, R., Holloway, J., Stutz, J., Petetin, H., McKeen, S., Beekmann, M., Warneke, C., Parrish, D. D., and de Gouw, J. A.: Emission ratios of anthropogenic volatile organic compounds in northern mid-latitude megacities: Observations versus emission inventories in Los Angeles and Paris, J. Geophys. Res.-Atmos., 118, 2041–2057, https://doi.org/10.1002/jgrd.50059, 2013.
Bruns, E. A., El Haddad, I., Keller, A., Klein, F., Kumar, N. K., Pieber, S. M., Corbin, J. C., Slowik, J. G., Brune, W. H., Baltensperger, U., and Prévôt, A. S. H.: Inter-comparison of laboratory smog chamber and flow reactor systems on organic aerosol yield and composition, Atmos. Meas. Tech., 8, 2315–2332, https://doi.org/10.5194/amt-8-2315-2015, 2015.
Bruns, E. A., El Haddad, I., Slowik, J. G., Kilic, D., Klein, F., Baltensperger, U., and Prévôt, A. S. H.: Identification of significant precursor gases of secondary organic aerosols from residential wood combustion, Sci. Rep., 6, 27881, https://doi.org/10.1038/srep27881, 2016.
Buhr, K., van Ruth, S., and Delahunty, C.: Analysis of volatile flavour compounds by Proton Transfer Reaction-Mass Spectrometry: fragmentation patterns and discrimination between isobaric and isomeric compounds, Int. J. Mass Spectrom., 221, 1–7, https://doi.org/10.1016/S1387-3806(02)00896-5, 2002.
Calderon-Garciduenas, L. and Villarreal-Rios, R.: Living close to heavy traffic roads, air pollution, and dementia, Lancet, 389, 675–677, https://doi.org/10.1016/S0140-6736(16)32596-X, 2017.
Canagaratna, M. R., Jayne, J. T., Jimenez, J. L., Allan, J. D., Alfarra, M. R., Zhang, Q., Onasch, T. B., Drewnick, F., Coe, H., and Middlebrook, A.: Chemical and microphysical characterization of ambient aerosols with the Aerodyne aerosol mass spectrometer, Mass Spectrom. Rev., 26, 185–222, 2007.
Cappellin, L., Probst, M., Limtrakul, J., Biasioli, F., Schuhfried, E., Soukoulis, C., Märk, T. D., and Gasperi, F.: Proton transfer reaction rate coefficients between H3O+ and some sulphur compounds, Int. J. Mass Spectrom., 295, 43–48, https://doi.org/10.1016/j.ijms.2010.06.023, 2010.
Cappellin, L., Karl, T., Probst, M., Ismailova, O., Winkler, P. M., Soukoulis, C., Aprea, E., Märk, T. D., Gasperi, F., and Biasioli, F.: On quantitative determination of volatile organic compound concentrations using proton transfer reaction time-of-flight mass spectrometry, Environ. Sci. Technol., 46, 2283–2290, https://doi.org/10.1021/es203985t, 2012.
Chan, T. W., Meloche, E., Kubsh, J., and Brezny, R.: Black carbon emissions in gasoline exhaust and a reduction alternative with a gasoline particulate filter, Environ. Sci. Technol., 48, 6027–6034, https://doi.org/10.1021/es501791b, 2014.
Czerwinski, J., Comte, P., Heeb, N., Mayer, A., and Hensel, V.: Nanoparticle Emissions of DI Gasoline Cars with/without GPF, SAE Technical Paper 2017-01-1004, https://doi.org/10.1021/acs.est.7b05045, 2017.
De Gouw, J. and Warneke, C.: Measurements of volatile organic compounds in the earth's atmosphere using proton-transfer-reaction mass spectrometry, Mass Spectrom. Rev., 26, 223–257, https://doi.org/10.1002/mas.20119, 2007.
de Gouw, J. A., Goldan, P. D., Warneke, C., Kuster, W. C., Roberts, J. M., Marchewka, M., Bertman, S. B., Pszenny, A. A. P., and Keene, W. C.: Validation of proton transfer reaction-mass spectrometry (PTR-MS) measurements of gas-phase organic compounds in the atmosphere during the New England Air Quality Study (NEAQS) in 2002, J. Geophys. Res.-Atmos., 108, 4682, https://doi.org/10.1029/2003jd003863, 2003.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
Demuynck, J. F. C., Bosteels, D., Hamje, H., and Anderson, J.: Real-World Emissions Measurements of a Gasoline Direct Injection Vehicle without and with a Gasoline Particulate Filter, SAE Technical Paper 2017-01-0985, https://doi.org/10.4271/2017-01-0985, 2017.
di Rattalma, M. F. and Perotti, G.: The Dieselgate: A Legal Perspective, Springer International Publishing AG, 179–218, available at: https://doi.org/10.1007/978-3-319-48323-8_11, 2017.
Donahue, N. M., Robinson, A. L., Stanier, C. O., and Pandis, S. N.: Coupled partitioning, dilution, and chemical aging of semivolatile organics, Environ. Sci. Technol., 40, 2635–2643, 2006.
Drinovec, L., Mocnik, G., Zotter, P., Prévôt, A. S. H., Ruckstuhl, C., Coz, E., Rupakheti, M., Sciare, J., Müller, T., Wiedensohler, A., and Hansen, A. D. A.: The “dual-spot” Aethalometer: an improved measurement of aerosol black carbon with real-time loading compensation, Atmos. Meas. Tech., 8, 1965–1979, https://doi.org/10.5194/amt-8-1965-2015, 2015.
Erickson, M. H., Gueneron, M., and Jobson, B. T.: Measuring long chain alkanes in diesel engine exhaust by thermal desorption PTR-MS, Atmos. Meas. Tech., 7, 225–239, https://doi.org/10.5194/amt-7-225-2014, 2014.
Finlayson-Pitts, B. J. and Pitts, J. N.: Chemistry of the Upper and Lower Atmosphere, Theory, Experiments, and Applications, Academic Press, San Diego, 969 pp., 2000.
Gentner, D. R., Jathar, S. H., Gordon, T. D., Bahreini, R., Day, D. A., El Haddad, I., Hayes, P. L., Pieber, S. M., Platt, S. M., de Gouw, J., Goldstein, A. H., Harley, R. A., Jimenez, J. L., Prévôt, A. S., and Robinson, A. L.: Review of Urban Secondary Organic Aerosol Formation from Gasoline and Diesel Motor Vehicle Emissions, Environ. Sci. Technol., 51, 1074–1093, https://doi.org/10.1021/acs.est.6b04509, 2017.
Gordon, T. D., Tkacik, D. S., Presto, A. A., Zhang, M., Jathar, S., Nguyen, N., Massetti, J., Truong, T., Cicero-Fernandez, P., Maddox, C., Rieger, P., Chattopadhyay, S., Maldonado, H., Maricq, M., M., and Robinson, A. L.: Primary gas-and particle-phase emissions and secondary organic aerosol production from gasoline and diesel off-road engines, Environ. Sci. Technol., 47, 1413–14146, 2013.
Gordon, T. D., Presto, A. A., May, A. A., Nguyen, N. T., Lipsky, E. M., Donahue, N. M., Gutierrez, A., Zhang, M., Maddox, C., Rieger, P., Chattopadhyay, S., Maldonado, H., Maricq, M. M., and Robinson, A. L.: Secondary organic aerosol formation exceeds primary particulate matter emissions for light-duty gasoline vehicles, Atmos. Chem. Phys., 14, 4661–4678, https://doi.org/10.5194/acp-14-4661-2014, 2014a.
Gordon, T. D., Presto, A. A., Nguyen, N. T., Robertson, W. H., Na, K., Sahay, K. N., Zhang, M., Maddox, C., Rieger, P., Chattopadhyay, S., Maldonado, H., Maricq, M. M., and Robinson, A. L.: Secondary organic aerosol production from diesel vehicle exhaust: impact of aftertreatment, fuel chemistry and driving cycle, Atmos. Chem. Phys., 14, 4643–4659, https://doi.org/10.5194/acp-14-4643-2014, 2014b.
Graus, M., Muller, M., and Hansel, A.: High resolution PTR-TOF: Quantification and formula confirmation of VOC in real time, J. Am. Soc. Mass. Spectr., 21, 1037–1044, 2010.
Gueneron, M., Erickson, M. H., Vanderschelden, G. S., and Jobson, B. T.: PTR-MS fragmentation patterns of gasoline hydrocarbons, Int. J. Mass Spectrom., 379, 97–109, https://doi.org/10.1016/j.ijms.2015.01.001, 2015.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Heeb, N. V., Forss, A. M., Brühlmann, S., Lüscher, R., Saxer, C. J., and Hug, P.: Three-way catalyst-induced formation of ammonia-velocity- and acceleration-dependent emission factors, Atmos. Environ., 40, 5986–5997, https://doi.org/10.1016/j.atmosenv.2005.12.035, 2006.
Hildebrandt, L., Donahue, N. M., and Pandis, S. N.: High formation of secondary organic aerosol from the photo-oxidation of toluene, Atmos. Chem. Phys., 9, 2973–2986, https://doi.org/10.5194/acp-9-2973-2009, 2009.
Jathar, S. H., Gordon, T. D., Hennigan, C. J., Pye, H. O. T., Pouliot, G., Adams, P. J., Donahue, N. M., and Robinson, A. L.: Unspeciated organic emissions from combustion sources and their influence on the secondary organic aerosol budget in the United States, P. Natl. Acad. Sci. USA, 111, 10473–10478, https://doi.org/10.1073/pnas.1323740111, 2014.
Jordan, A., Haidacher, S., Hanel, G., Hartungen, E., Märk, L., Seehauser, H., Schottkowsky, R., Sulzer, P., and Märk, T. D.: A high resolution and high sensitivity proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS), Int. J. Mass Spectrom., 286, 122–128, https://doi.org/10.1016/j.ijms.2009.07.005, 2009.
Jordan, A., Jaksch, S., Jürschik, S., Edtbauer, A., Agarwal, B., Hanel, G., Hartungen, E., Seehauser, H., Märk, L., Sulzer, P., and Märk, T. D.: H3O+, NO+ and as precursor ions in PTR-MS: isomeric VOC compounds and reactions with different chemical groups, 5th International Conference on Proton Transfer Reaction Mass Spectrometry and its Applications, 2011.
Jordan, C. E., Ziemann, P. J., Griffin, R. J., Lim, Y. B., Atkinson, R., and Arey, J.: Modeling SOA formation from OH reactions with C8–C17 n-alkanes, Atmos. Environ., 42, 8015–8026, https://doi.org/10.1016/j.atmosenv.2008.06.017, 2008.
Kang, E., Root, M. J., Toohey, D. W., and Brune, W. H.: Introducing the concept of Potential Aerosol Mass (PAM), Atmos. Chem. Phys., 7, 5727–5744, https://doi.org/10.5194/acp-7-5727-2007, 2007.
Karjalainen, P., Pirjola, L., Heikkilä, J., Lähde, T., Tzamkiozis, T., Ntziachristos, L., Keskinen, J., and Rönkkö, T.: Exhaust particles of modern gasoline vehicles: A laboratory and an on-road study, Atmos. Environ., 97, 262–270, https://doi.org/10.1016/j.atmosenv.2014.08.025, 2014.
Karjalainen, P., Timonen, H., Saukko, E., Kuuluvainen, H., Saarikoski, S., Aakko-Saksa, P., Murtonen, T., Bloss, M., Dal Maso, M., Simonen, P., Ahlberg, E., Svenningsson, B., Brune, W. H., Hillamo, R., Keskinen, J., and Rönkkö, T.: Time-resolved characterization of primary particle emissions and secondary particle formation from a modern gasoline passenger car, Atmos. Chem. Phys., 16, 8559–8570, https://doi.org/10.5194/acp-16-8559-2016, 2016.
Knighton, W. B., Fortner, E. C., Midey, A. J., Viggiano, A. A., Herndon, S. C., Wood, E. C., and Kolb, C. E.: HCN detection with a proton transfer reaction mass spectrometer, Int. J. Mass Spectrom., 283, 112–121, https://doi.org/10.1016/j.ijms.2009.02.013, 2009.
Krechmer, J. E., Pagonis, D., Ziemann, P. J., and Jimenez, J. L.: Quantification of Gas-Wall Partitioning in Teflon Environmental Chambers Using Rapid Bursts of Low-Volatility Oxidized Species Generated in Situ, Environ. Sci. Technol., 50, 5757–5765, https://doi.org/10.1021/acs.est.6b00606, 2016.
Kunzi, L., Krapf, M., Daher, N., Dommen, J., Jeannet, N., Schneider, S., Platt, S., Slowik, J. G., Baumlin, N., Salathe, M., Prévôt, A. S. H., Kalberer, M., Strahl, C., Dumbgen, L., Sioutas, C., Baltensperger, U., and Geiser, M.: Toxicity of aged gasoline exhaust particles to normal and diseased airway epithelia, Sci. Rep., 5, 11801, https://doi.org/10.1038/srep11801, 2015.
La, Y. S., Camredon, M., Ziemann, P. J., Valorso, R., Matsunaga, A., Lannuque, V., Lee-Taylor, J., Hodzic, A., Madronich, S., and Aumont, B.: Impact of chamber wall loss of gaseous organic compounds on secondary organic aerosol formation: explicit modeling of SOA formation from alkane and alkene oxidation, Atmos. Chem. Phys., 16, 1417–1431, https://doi.org/10.5194/acp-16-1417-2016, 2016.
Lambe, A., Massoli, P., Zhang, X., Canagaratna, M., Nowak, J., Daube, C., Yan, C., Nie, W., Onasch, T., Jayne, J., Kolb, C., Davidovits, P., Worsnop, D., and Brune, W.: Controlled nitric oxide production via O(1D) + N2O reactions for use in oxidation flow reactor studies, Atmos. Meas. Tech., 10, 2283–2298, https://doi.org/10.5194/amt-10-2283-2017, 2017.
Lambe, A. T., Ahern, A. T., Williams, L. R., Slowik, J. G., Wong, J. P. S., Abbatt, J. P. D., Brune, W. H., Ng, N. L., Wright, J. P., Croasdale, D. R., Worsnop, D. R., Davidovits, P., and Onasch, T. B.: Characterization of aerosol photooxidation flow reactors: heterogeneous oxidation, secondary organic aerosol formation and cloud condensation nuclei activity measurements, Atmos. Meas. Tech., 4, 445–461, https://doi.org/10.5194/amt-4-445-2011, 2011.
Lambe, A. T., Chhabra, P. S., Onasch, T. B., Brune, W. H., Hunter, J. F., Kroll, J. H., Cummings, M. J., Brogan, J. F., Parmar, Y., Worsnop, D. R., Kolb, C. E., and Davidovits, P.: Effect of oxidant concentration, exposure time, and seed particles on secondary organic aerosol chemical composition and yield, Atmos. Chem. Phys., 15, 3063–3075, https://doi.org/10.5194/acp-15-3063-2015, 2015.
Li, L., Tang, P., Nakao, S., Chen, C.-L., and Cocker III, D. R.: Role of methyl group number on SOA formation from monocyclic aromatic hydrocarbons photooxidation under low-NOx conditions, Atmos. Chem. Phys., 16, 2255–2272, https://doi.org/10.5194/acp-16-2255-2016, 2016a.
Li, L., Tang, P., Nakao, S., and Cocker III, D. R.: Impact of molecular structure on secondary organic aerosol formation from aromatic hydrocarbon photooxidation under low-NOx conditions, Atmos. Chem. Phys., 16, 10793–10808, https://doi.org/10.5194/acp-16-10793-2016, 2016b.
Li, L., Qi, L., and Cocker, D. R.: Contribution of methyl group to secondary organic aerosol formation from aromatic hydrocarbon photooxidation, Atmos. Environ., 151, 133–139, https://doi.org/10.1016/j.atmosenv.2016.11.064, 2017.
Li, R., Palm, B. B., Ortega, A. M., Hlywiak, J., Hu, W., Peng, Z., Day, D. A., Knote, C., Brune, W. H., de Gouw, J. A., and Jimenez, J. L.: Modeling the radical chemistry in an oxidation flow reactor: radical formation and recycling, sensitivities, and the OH exposure estimation equation, J. Phys. Chem. A., 119, 4418–4432, https://doi.org/10.1021/jp509534k, 2015.
Lim, Y. B. and Ziemann, P. J.: Products and mechanism of secondary organic aerosol formation from reactions of n-alkanes with OH radicals in the presence of NOx, Environ. Sci. Technol., 39, 9229–9236, https://doi.org/10.1021/es051447g, 2005.
Lindinger, W. and Jordan, A.: Proton-transfer-reaction mass spectrometry (PTR-MS): on-line monitoring of volatile organic compounds at pptv levels, Chem. Soc. Rev., 27, 347–375, https://doi.org/10.1039/A827347Z, 1998.
Loza, C. L., Chhabra, P. S., Yee, L. D., Craven, J. S., Flagan, R. C., and Seinfeld, J. H.: Chemical aging of m-xylene secondary organic aerosol: laboratory chamber study, Atmos. Chem. Phys., 12, 151–167, https://doi.org/10.5194/acp-12-151-2012, 2012.
Loza, C. L., Craven, J. S., Yee, L. D., Coggon, M. M., Schwantes, R. H., Shiraiwa, M., Zhang, X., Schilling, K. A., Ng, N. L., Canagaratna, M. R., Ziemann, P. J., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol yields of 12-carbon alkanes, Atmos. Chem. Phys., 14, 1423–1439, https://doi.org/10.5194/acp-14-1423-2014, 2014.
May, A. A., Nguyen, N. T., Presto, A. A., Gordon, T. D., Lipsky, E. M., Karve, M., Gutierrez, A., Robertson, W. H., Zhang, M., Brandow, C., Chang, O., Chen, S., Cicero-Fernandez, P., Dinkins, L., Fuentes, M., Huang, S. M., Ling, R., Long, J., Maddox, C., Massetti, J., McCauley, E., Miguel, A., Na, K., Ong, R., Pang, Y., Rieger, P., Sax, T., Truong, T., Vo, T., Chattopadhyay, S., Maldonado, H., Maricq, M. M., and Robinson, A. L.: Gas- and particle-phase primary emissions from in-use, on-road gasoline and diesel vehicles, Atmos. Environ., 88, 247–260, https://doi.org/10.1016/j.atmosenv.2014.01.046, 2014.
McMurry, P. H. and Grosjean, D.: Gas and aerosol wall losses in Teflon film smog chambers, Environ. Sci. Technol., 19, 1176–1182, https://doi.org/10.1021/es00142a006, 1985.
Molteni, U., Bianchi, F., Klein, F., El Haddad, I., Frege, C., Rossi, M. J., Dommen, J., and Baltensperger, U.: Formation of highly oxygenated organic molecules from aromatic compounds, Atmos. Chem. Phys., 18, 1909–1921, https://doi.org/10.5194/acp-18-1909-2018, 2018.
Müller, M., Mikoviny, T., and Wisthaler, A.: Detector aging induced mass discrimination and non-linearity effects in PTR-ToF-MS, Int. J. Mass Spectrom., 365–366, 93–97, https://doi.org/10.1016/j.ijms.2013.12.008, 2014.
Muñoz, M., Haag, R., Honegger, P., Zeyer, K., Mohn, J., Comte, P., Czerwinski, J., and Heeb, N. V.: Co-formation and co-release of genotoxic PAHs, alkyl-PAHs and soot nanoparticles from gasoline direct injection vehicles, Atmos. Environ., 178, 242–254, https://doi.org/10.1016/j.atmosenv.2018.01.050, 2018.
Ng, N. L., Chhabra, P. S., Chan, A. W. H., Surratt, J. D., Kroll, J. H., Kwan, A. J., McCabe, D. C., Wennberg, P. O., Sorooshian, A., Murphy, S. M., Dalleska, N. F., Flagan, R. C., and Seinfeld, J. H.: Effect of NOx level on secondary organic aerosol (SOA) formation from the photooxidation of terpenes, Atmos. Chem. Phys., 7, 5159–5174, https://doi.org/10.5194/acp-7-5159-2007, 2007a.
Ng, N. L., Kroll, J. H., Chan, A. W. H., Chhabra, P. S., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from m-xylene, toluene, and benzene, Atmos. Chem. Phys., 7, 3909–3922, https://doi.org/10.5194/acp-7-3909-2007, 2007b.
Ng, N. L., Canagaratna, M. R., Jimenez, J. L., Chhabra, P. S., Seinfeld, J. H., and Worsnop, D. R.: Changes in organic aerosol composition with aging inferred from aerosol mass spectra, Atmos. Chem. Phys., 11, 6465–6474, https://doi.org/10.5194/acp-11-6465-2011, 2011.
Nordin, E. Z., Eriksson, A. C., Roldin, P., Nilsson, P. T., Carlsson, J. E., Kajos, M. K., Hellén, H., Wittbom, C., Rissler, J., Löndahl, J., Swietlicki, E., Svenningsson, B., Bohgard, M., Kulmala, M., Hallquist, M., and Pagels, J. H.: Secondary organic aerosol formation from idling gasoline passenger vehicle emissions investigated in a smog chamber, Atmos. Chem. Phys., 13, 6101–6116, https://doi.org/10.5194/acp-13-6101-2013, 2013.
Odum, J. R., Jungkamp, T. P. W., Griffin, R. J., Forstner, H. J. L., Flagan, R. C., and Seinfeld, J. H.: Aromatics, reformulated gasoline, and atmospheric organic aerosol formation, Environ. Sci. Technol., 31, 1890–1897, 1997.
Ortega, A. M., Day, D. A., Cubison, M. J., Brune, W. H., Bon, D., de Gouw, J. A., and Jimenez, J. L.: Secondary organic aerosol formation and primary organic aerosol oxidation from biomass-burning smoke in a flow reactor during FLAME-3, Atmos. Chem. Phys., 13, 11551–11571, https://doi.org/10.5194/acp-13-11551-2013, 2013.
Pagonis, D., Krechmer, J. E., de Gouw, J., Jimenez, J. L., and Ziemann, P. J.: Effects of gas-wall partitioning in Teflon tubing and instrumentation on time-resolved measurements of gas-phase organic compounds, Atmos. Meas. Tech., 10, 4687–4696, https://doi.org/10.5194/amt-10-4687-2017, 2017.
Palm, B. B., Campuzano-Jost, P., Ortega, A. M., Day, D. A., Kaser, L., Jud, W., Karl, T., Hansel, A., Hunter, J. F., Cross, E. S., Kroll, J. H., Peng, Z., Brune, W. H., and Jimenez, J. L.: In situ secondary organic aerosol formation from ambient pine forest air using an oxidation flow reactor, Atmos. Chem. Phys., 16, 2943–2970, https://doi.org/10.5194/acp-16-2943-2016, 2016.
Pandis, S. N., Donahue, N. M., Murphy, B. N., Riipinen, I., Fountoukis, C., Karnezi, E., Patoulias, D., and Skyllakou, K.: Introductory lecture: atmospheric organic aerosols: insights from the combination of measurements and chemical transport models, Faraday Discuss., 165, 9–24, https://doi.org/10.1039/C3FD00108C, 2013.
Pankow, J. F.: An Absorption-Model of Gas-Particle Partitioning of Organic-Compounds in the Atmosphere, Atmos. Environ., 28, 185–188, https://doi.org/10.1016/1352-2310(94)90093-0, 1994.
Peng, Z. and Jimenez, J. L.: Modeling of the chemistry in oxidation flow reactors with high initial NO, Atmos. Chem. Phys., 17, 11991–12010, https://doi.org/10.5194/acp-17-11991-2017, 2017.
Peng, Z., Day, D. A., Stark, H., Li, R., Lee-Taylor, J., Palm, B. B., Brune, W. H., and Jimenez, J. L.: HOx radical chemistry in oxidation flow reactors with low-pressure mercury lamps systematically examined by modeling, Atmos. Meas. Tech., 8, 4863–4890, https://doi.org/10.5194/amt-8-4863-2015, 2015.
Peng, Z., Day, D. A., Ortega, A. M., Palm, B. B., Hu, W., Stark, H., Li, R., Tsigaridis, K., Brune, W. H., and Jimenez, J. L.: Non-OH chemistry in oxidation flow reactors for the study of atmospheric chemistry systematically examined by modeling, Atmos. Chem. Phys., 16, 4283–4305, https://doi.org/10.5194/acp-16-4283-2016, 2016.
Pieber, S. M., El Haddad, I., Slowik, J. G., Canagaratna, M. R., Jayne, J. T., Platt, S. M., Bozzetti, C., Daellenbach, K. R., Frohlich, R., Vlachou, A., Klein, F., Dommen, J., Miljevic, B., Jimenez, J. L., Worsnop, D. R., Baltensperger, U., and Prévôt, A. S. H.: Inorganic Salt Interference on in Aerodyne AMS and ACSM Organic Aerosol Composition Studies, Environ. Sci. Technol., 50, 10494–10503, 10.1021/acs.est.6b01035, 2016.
Pieber S. and Prévôt A.: Pieber_ACP2018_GasolineSOA Data, available at: https://doi.org/10.5281/zenodo.1305770 (last access: 10 July 2018), 2018.
Platt, S. M., El Haddad, I., Zardini, A. A., Clairotte, M., Astorga, C., Wolf, R., Slowik, J. G., Temime-Roussel, B., Marchand, N., Ježek, I., Drinovec, L., Močnik, G., Möhler, O., Richter, R., Barmet, P., Bianchi, F., Baltensperger, U., and Prévôt, A. S. H.: Secondary organic aerosol formation from gasoline vehicle emissions in a new mobile environmental reaction chamber, Atmos. Chem. Phys., 13, 9141–9158, https://doi.org/10.5194/acp-13-9141-2013, 2013.
Platt, S. M., El Haddad, I., Pieber, S. M., Huang, R. J., Zardini, A. A., Clairotte, M., Suarez-Bertoa, R., Barmet, P., Pfaffenberger, L., Wolf, R., Slowik, J. G., Fuller, S. J., Kalberer, M., Chirico, R., Dommen, J., Astorga, C., Zimmermann, R., Marchand, N., Hellebust, S., Temime-Roussel, B., Baltensperger, U., and Prévôt, A. S.: Two-stroke scooters are a dominant source of air pollution in many cities, Nat. Commun., 5, 3749, https://doi.org/10.1038/ncomms4749, 2014.
Platt, S. M., El Haddad, I., Pieber, S. M., Zardini, A. A., Suarez-Bertoa, R., Clairotte, M., Daellenbach, K. R., Huang, R. J., Slowik, J. G., Hellebust, S., Temime-Roussel, B., Marchand, N., de Gouw, J., Jimenez, J. L., Hayes, P. L., Robinson, A. L., Baltensperger, U., Astorga, C., and Prévôt, A. S. H.: Gasoline cars produce more carbonaceous particulate matter than modern filter-equipped diesel cars, Sci. Rep., 7, 4926, https://doi.org/10.1038/s41598-017-03714-9, 2017.
Saliba, G., Saleh, R., Zhao, Y., Presto, A. A., Lambe, A. T., Frodin, B., Sardar, S., Maldonado, H., Maddox, C., May, A. A., Drozd, G. T., Goldstein, A. H., Russell, L. M., Hagen, F. P., and Robinson, A. L.: A comparison of gasoline direct injection (GDI) and port fuel injection (PFI) vehicle emissions: emission certification standards, cold start, secondary organic aerosol formation potential, and potential climate impacts, Environ. Sci. Technol., 51, 6542–6552, https://doi.org/10.1021/acs.est.6b06509, 2017.
Schauer, J. J., Kleeman, M. J., Cass, G. R., and Simoneit, B. R. T.: Measurement of emissions from air pollution sources. 2. C1 through C30 organic compounds from medium duty diesel trucks, Environ. Sci. Technol., 33, 1578–1587, 1999.
Schauer, J. J., Kleeman, M. J., Cass, G. R., and Simoneit, B. R. T.: Measurement of emissions from air pollution sources. 5. C1-C32 organic compounds from gasoline-powered motor vehicles, Environ. Sci. Technol., 36, 1169–1180, 2002.
Schwantes, R. H., Schilling, K. A., McVay, R. C., Lignell, H., Coggon, M. M., Zhang, X., Wennberg, P. O., and Seinfeld, J. H.: Formation of highly oxygenated low-volatility products from cresol oxidation, Atmos. Chem. Phys., 17, 3453–3474, https://doi.org/10.5194/acp-17-3453-2017, 2017.
Stirnweis, L., Marcolli, C., Dommen, J., Barmet, P., Frege, C., Platt, S. M., Bruns, E. A., Krapf, M., Slowik, J. G., Wolf, R., Prévôt, A. S. H., Baltensperger, U., and El-Haddad, I.: Assessing the influence of NOx concentrations and relative humidity on secondary organic aerosol yields from α-pinene photo-oxidation through smog chamber experiments and modelling calculations, Atmos. Chem. Phys., 17, 5035–5061, https://doi.org/10.5194/acp-17-5035-2017, 2017.
Suarez-Bertoa, R., Zardini, A. A., and Astorga, C.: Ammonia exhaust emissions from spark ignition vehicles over the New European Driving Cycle, Atmos. Environ., 97, 43–53, https://doi.org/10.1016/j.atmosenv.2014.07.050, 2014.
Taira, M. and Kanda, Y.: Continuous generation system for low-concentration gaseous nitrous acid, Anal. Chem., 62, 630–633, 1990.
Tkacik, D. S., Lambe, A. T., Jathar, S., Li, X., Presto, A. A., Zhao, Y., Blake, D., Meinardi, S., Jayne, J. T., Croteau, P. L., and Robinson, A. L.: Secondary organic aerosol formation from in-use motor vehicle emissions using a potential aerosol mass reactor, Environ. Sci. Technol., 48, 11235–11242, https://doi.org/10.1021/es502239v, 2014.
Turpin, B. J. and Lim, H.-J.: Species Contributions to PM2.5 Mass Concentrations: Revisiting Common Assumptions for Estimating Organic Mass, Aerosol Sci. Technol., 35, 602–610, https://doi.org/10.1080/02786820119445, 2001.
Wang, T., Jerrett, M., Sinsheimer, P., and Zhu, Y.: Estimating PM2.5-associated mortality increase in California due to the Volkswagen emission control defeat device, Atmos. Environ., 144, 168–174, https://doi.org/10.1016/j.atmosenv.2016.08.074, 2016.
Weitkamp, E. A., Sage, A. M., Pierce, J. R., Donahue, N. M., and Robinson, A. L.: Organic aerosol formation from photochemical oxidation of diesel exhaust in a smog chamber, Environ. Sci. Technol., 41, 6969–6975, https://doi.org/10.1021/es070193r, 2007.
Worton, D. R., Isaacman, G., Gentner, D. R., Dallmann, T. R., Chan, A. W., Ruehl, C., Kirchstetter, T. W., Wilson, K. R., Harley, R. A., and Goldstein, A. H.: Lubricating oil dominates primary organic aerosol emissions from motor vehicles, Environ. Sci. Technol., 48, 3698–3706, https://doi.org/10.1021/es405375j, 2014.
Ye, P., Ding, X., Hakala, J., Hofbauer, V., Robinson, E. S., and Donahue, N. M.: Vapor wall loss of semi-volatile organic compounds in a Teflon chamber, Aerosol Sci. Technol., 50, 822–834, https://doi.org/10.1080/02786826.2016.1195905, 2016.
Zhang, X., Cappa, C. D., Jathar, S. H., McVay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, https://doi.org/10.1073/pnas.1404727111, 2014.
Zhao, Y., Nguyen, N. T., Presto, A. A., Hennigan, C. J., May, A. A., and Robinson, A. L.: Intermediate Volatility Organic Compound Emissions from On-Road Gasoline Vehicles and Small Off-Road Gasoline Engines, Environ. Sci. Technol., 50, 4554–4563, https://doi.org/10.1021/acs.est.5b06247, 2016.
Zhao, Y., Saleh, R., Saliba, G., Presto, A. A., Gordon, T. D., Drozd, G. T., Goldstein, A. H., Donahue, N. M., and Robinson, A. L.: Reducing secondary organic aerosol formation from gasoline vehicle exhaust, P. Natl. Acad. Sci. USA, 114, 6984–6989, https://doi.org/10.1073/pnas.1620911114, 2017.
Zhao, Y., Lambe, A. T., Saleh, R., Saliba, G., and Robinson, A. L.: Secondary Organic Aerosol Production from Gasoline Vehicle Exhaust: Effects of Engine Technology, Cold Start, and Emission Certification Standard, Environ. Sci. Technol., 52, 1253–1261, https://doi.org/10.1021/acs.est.7b05045, 2018.
Zhu, R., Hu, J., Bao, X., He, L., Lai, Y., Zu, L., Li, Y., and Su, S.: Tailpipe emissions from gasoline direct injection (GDI) and port fuel injection (PFI) vehicles at both low and high ambient temperatures, Environ. Pollut., 216, 223–234, https://doi.org/10.1016/j.envpol.2016.05.066, 2016.
Zimmerman, N., Wang, J. M., Jeong, C. H., Ramos, M., Hilker, N., Healy, R. M., Sabaliauskas, K., Wallace, J. S., and Evans, G. J.: Field Measurements of Gasoline Direct Injection Emission Factors: Spatial and Seasonal Variability, Environ. Sci. Technol., 50, 2035–2043, https://doi.org/10.1021/acs.est.5b04444, 2016a.
Zimmerman, N., Wang, J. M., Jeong, C. H., Wallace, J. S., and Evans, G. J.: Assessing the Climate Trade-Offs of Gasoline Direct Injection Engines, Environ. Sci. Technol., 50, 8385–8392, https://doi.org/10.1021/acs.est.6b01800, 2016b.
Zotter, P., El Haddad, I., Zhang, Y., Hayes, P. L., Zhang, X., Lin, Y.-H., Wacker, L., Schnelle-Kreis, J., Abbaszade, G., Zimmermann, R., Surratt, J. D., Weber, R., Jimenez, J. L., Szidat, S., Baltensperger, U., and Prévôt, A. S. H.: Diurnal cycle of fossil and nonfossil carbon using radiocarbon analyses during CalNex, J. Geophys. Res.-Atmos., 119, 6818–6835, https://doi.org/10.1002/2013jd021114, 2014.