the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 13 Jun 2018
| 13 Jun 2018
Ozone response to emission reductions in the southeastern United States
Charles L. Blanchard
George M. Hidy
Ozone (O3) formation in the southeastern US is studied in relation to nitrogen oxide (NOx) emissions using long-term (1990s–2015) surface measurements of the Southeastern Aerosol Research and Characterization (SEARCH) network, U.S. Environmental Protection Agency (EPA) O3 measurements, and EPA Clean Air Status and Trends Network (CASTNET) nitrate deposition data. Annual fourth-highest daily peak 8 h O3 mixing ratios at EPA monitoring sites in Georgia, Alabama, and Mississippi exhibit statistically significant (p < 0.0001) linear correlations with annual NOx emissions in those states between 1996 and 2015. The annual fourth-highest daily peak 8 h O3 mixing ratios declined toward values of ∼ 45–50 ppbv and monthly O3 maxima decreased at rates averaging ∼ 1–1.5 ppbv yr−1. Mean annual total oxidized nitrogen (NOy) mixing ratios at SEARCH sites declined in proportion to NOx emission reductions. CASTNET data show declining wet and dry nitrate deposition since the late 1990s, with total (wet plus dry) nitrate deposition fluxes decreasing linearly in proportion to reductions of NOx emissions by ∼ 60 % in Alabama and Georgia. Annual nitrate deposition rates at Georgia and Alabama CASTNET sites correspond to 30 % of Georgia emission rates and 36 % of Alabama emission rates, respectively. The fraction of NOx emissions lost to deposition has not changed. SEARCH and CASTNET sites exhibit downward trends in mean annual nitric acid (HNO3) concentrations. Observed relationships of O3 to NOz (NOy–NOx) support past model predictions of increases in cycling of NO and increasing responsiveness of O3 to NOx. The study data provide a long-term record that can be used to examine the accuracy of process relationships embedded in modeling efforts. Quantifying observed O3 trends and relating them to reductions in ambient NOy species concentrations offers key insights into processes of general relevance to air quality management and provides important information supporting strategies for reducing O3 mixing ratios.
- Article
(2864 KB) -
Supplement
(3010 KB) - BibTeX
- EndNote





Ozone (O3) is a well known and important product of photochemical processes in the troposphere involving nitric oxide (NO), nitrogen dioxide (NO2), and volatile organic compounds (VOCs). Ozone is of broad interest for its adverse effects on humans and ecosystems, as reflected by regulation through the US Clean Air Act (e.g., U.S. EPA, 2014, 2015a).
Regulatory actions address extreme O3 mixing ratios: the US National Ambient Air Quality Standard (NAAQS), currently 70 ppbv, is applicable to the annual fourth-highest daily 8 h maxima averaged over 3-year periods (U.S. EPA, 2015b, c). By the early 1990s, US emission control efforts began to focus on nitrogen oxides (NOx = NO + NO2) in addition to VOCs (NRC, 1991). O3 management has generally relied on precursor reduction requirements estimated from models that integrate descriptions of nonlinear chemical and atmospheric processes (e.g., Seigneur and Dennis, 2011), and guidance has also derived from so-called “observation-based” models linking O3 and its precursors based on chemical reactions that are believed to drive ambient mixing ratios (e.g., NARSTO, 2000; Schere and Hidy, 2000).
Most of the work developing an observational basis for O3-precursor chemistry derives from field campaigns, sometimes focusing on urban conditions. Short-term data are available from aircraft flights, for example, or summer field measurements made at a variety of locations. Such studies usually are limited to a month or two of intense sampling. One example in the southern US is the 1990 ROSE experiment at Kinterbish, a rural, forested state park in western Alabama (Frost et al., 1998). This summer study of rural O3 at low anthropogenic VOC and low NOx mixing ratios provided important insights into rural O3 formation (Trainer et al., 2000). Other examples of short-term campaigns across the US and elsewhere are reviewed in Solomon et al. (2000). More recent field studies include New England in 2002 (e.g., Griffin et al., 2004; Kleinman et al., 2007), Texas in 2006 (e.g., Neuman et al., 2009), the mid-Atlantic region in 2011 (He et al., 2013), California in 2010 (Ryerson et al., 2013), Colorado in 2012 and 2014 (e.g., McDuffie et al., 2016), and the southeastern US in 2013 (e.g., Neuman et al., 2016; Warneke et al., 2016). These campaigns and accompanying analyses of O3 production and accumulation typically address summer, which historically has the strongest photochemical activity. However, strong photochemical O3 production can occur under special circumstances in winter (e.g., Schnell et al., 2009).
Accounting for an O3 background is important. O3 background is associated with biogenic influence, large-scale transport, or the potential influence of the upper atmosphere (e.g., stratospheric intrusions, especially during spring; Lin et al., 2012; Langford et al., 2015). The nature and magnitude of background O3 remain an active area of research in the US and Europe (Naja et al., 2003; Solberg et al., 2005; Ordóñez et al., 2007; Cristofanelli and Bonasoni, 2009; Arif and Abdullah, 2011; Zhang et al., 2011; Wilson et al., 2012). Hidy and Blanchard (2015) discuss definitions of continental and regional background O3. For this study, we adopt a definition of “background” that includes both the non-anthropogenic component and the southeastern regional component (Sect. 4.4.1).
Field studies have provided observational evidence of nonlinearity in O3–NOz relationships (e.g., Trainer et al., 1993, 1995; Kleinman et al., 1994; Hirsch et al., 1996; Frost et al., 1998; Kasibhatla et al., 1998; Nunnermacker et al., 1998; St. John et al., 1998; Sillman et al., 1998; Zaveri et al., 2003; Griffin et al., 2004; Travis et al., 2016). Long-term, post-1990s data are widely available for O3 and NO2 but detailed observations of total oxidized nitrogen (NOy) and VOC, and especially their component species, are typically lacking (e.g., Hidy and Blanchard, 2015). One of the longest records of urban and suburban data, comprising a series of short-term campaigns as well as continuous measurements, is from southern California. This region exemplifies a photochemically active urban regime. An analysis of multi-decadal (since the 1960s) data by Pollack et al. (2013) reveals how changes in atmospheric chemical reactions have contributed to the observed reductions of O3 in southern California since 1973. Long-term (more than one decade) measurements characterizing O3 and NOy relationships in both urban and rural conditions are less common.
The photochemical regime in the southeast represents humid subtropical conditions with urban emissions yielding elevated O3 levels superimposed on a general regional background (Chameides and Cowling, 1995). The EPA O3 and deposition data provide a regional basis for characterizing trends since the early 1980s (U.S. EPA, 2016a, b). In addition, the Southeastern Aerosol Research and Characterization (SEARCH) project (Hansen et al., 2003; Hidy et al., 2014) provides measurements that can be used to investigate changes in O3 production resulting from changes in anthropogenic emissions in the southeastern US. The SEARCH network of eight sites began with the Southeastern Oxidant Study (SOS) (Chameides and Cowling, 1995; Meagher et al., 1998) rural locations, which were near (1) Centreville, AL, ∼ 85 km southwest of Birmingham; (2) at Yorkville, GA, ∼ 60 km northwest of Atlanta, GA; and (3) at Oak Grove, MS, ∼ 40 km southeast of Hattiesburg, MS, and 75 km north of Gulfport, MS, on private land within the confines of the De Soto National Forest (Hansen et al., 2003). Measurements of some gas-phase species began at these rural sites in 1992, thus providing a rural data record of over 20 years. Beginning in 1999, SEARCH added five sites in metropolitan Atlanta, GA; Birmingham, AL; Pensacola, FL; and Gulfport, MS.
Our goal for this study is to extend earlier analyses of the photochemical response of O3 to precursors through 2014, emphasizing relationships between O3 and NOy. We first summarize relevant O3 photochemistry to provide a context for the observational analysis. We then describe trends in emissions and ambient pollutant concentrations, and discuss O3, NOz, and HNO3 observations at the SEARCH sites. The trends in O3 mixing ratio, NOx precursor emissions, and ambient nitrogen oxide mixing ratios offer important insight into future changes in O3 and NOy. Blanchard et al. (2014) previously explained the majority (66–80 %) of the day-to-day variations in daily peak 8 h average O3 at SEARCH sites during March–October of 2002–2011 using meteorological variables coupled with ambient measurements of O3 precursors (NO, NO2; limited measurements of VOCs) and NOx photochemical reaction products (NOz) and a statistical model. The previous analyses are extended here for data through 2014 to help understand ongoing and potential future O3 changes in relation to changes in ambient NOz and HNO3 mixing ratios in the southeastern US. Results are discussed in relation to modeling predictions by Reynolds et al. (2004) and others.
2.1 Key atmospheric reactions linking O3 with NOx
Net tropospheric O3 accumulation occurs when sunlight acts on VOC and NOx emissions and the O3 production rate exceeds O3 loss (Trainer et al., 2000). Tropospheric O3 mixing ratios are affected by solar intensity, chemical formation and loss (e.g., deposition) rates of O3, the rate of dispersion of O3 and its precursors, meteorological factors, and vertical entrainment and transport of plumes. NO2 forms rapidly by reaction of NO with O3 and photolysis of NO2 produces O3, yielding steady-state mixing ratios of NO, NO2, and O3 in the absence of other species as expressed by the photostationary state, or Leighton relationship (Seinfeld, 1986).
In the troposphere, NO2 also forms by reaction of NO with peroxy (HO2) and alkyl peroxy (RO2) free radical species, which derive in turn from the reaction of VOCs with hydroxyl (HO), HO2, RO2, and alkyl radicals (Seinfeld, 1986). Radical production from VOCs creates a pathway for conversion of NO to NO2 that does not consume O3 (Atkinson, 2000), which then leads to higher O3 mixing ratios.
O3 accumulation is typically associated with high solar radiation intensity and temperatures favoring atmospheric reactions, lower wind speeds, and high anthropogenic emission rates (NARSTO, 2000). O3 accumulation requires NO mixing ratios exceeding approximately 10 to 30 pptv (Atkinson, 2000; Logan, 1985), along with the presence of HO2 and RO2 radicals that react with NO to form NO2. The former conditions are normally met in urban air; NOx mixing ratios are much lower under typical conditions in rural southeastern areas, but still well above 30 pptv (e.g., Hudman et al., 2007; Travis et al., 2016). Under these conditions, the O3 photochemical production rate is proportional to the ambient NO multiplied by the sum of HO2 and RO2 radical mixing ratios, where the latter are weighted by their rates of reaction with NO (Trainer et al., 2000). Field studies show that observed rates of rural O3 production are proportional to the rate of oxidation of NOx. Where VOCs are present for radical production and NOx is rate limiting (Trainer et al., 2000), regional O3 production can be expressed in terms of the derivative d[O3]∕d[NOx], denoted as the O3 production efficiency (OPE) (Liu et al., 1987). OPE is understood as the number of molecules of O3 formed per molecule of NOx oxidized and OPE increases as NOx mixing ratios decrease (Liu et al., 1987; Trainer et al., 2000). OPE reflects the mean number of NO–NO2 cycles occurring, in which each photolysis of one NO2 molecule generates one O3 molecule until that NO2 molecule is oxidized to nitric acid (HNO3) or to other species such as peroxyacetyl nitrate (PAN). NOx reaction products, including HNO3 and PAN, comprise NOz. For chemical reactions, the quantity d[O3]∕d[NOz] is equivalent to d[O3]∕d[NOx], but with opposite sign, and has therefore been used to estimate OPE; limitations due to confounding influences of emissions, transport, and deposition are discussed in Sect. 4.3 and 4.4.
Empirically, the slope of a linear fit of afternoon O3 (or Ox = O3 + NO2) versus NOz has been used to estimate OPE (e.g., Trainer et al., 1993; Pollack et al., 2013). This estimate is subject to certain limitations because it does not explicitly account for (1) day-to-day variability in “old” (baseline or regional background) O3 mixing ratios, (2) mixing of air masses having different emission histories, (3) rapid loss of HNO3 (primarily through dry deposition, but also through gas-to-particle conversion) (Trainer et al., 2000), and (4) regeneration of NO2 from PAN and certain other species. Because PAN regenerates NO2, it can serve as a reservoir rather than a true NO2 sink (Singh and Hanst, 1981; Singh, 1987). In contrast, HNO3 largely terminates the cycling between NO and NO2. Therefore, the relative yields of PAN and HNO3 are of importance. Despite such limitations in using measurements to quantify OPE, data from field studies have been used since the 1990s to determine upper bounds for OPE and the results have continued to appear in the literature as an indicator of relevance to O3 chemistry (e.g., Berkowitz et al., 2004; Neuman et al., 2009; Kim et al., 2016). Investigators caution that field measurements reveal the net of production and loss, which potentially overestimates actual OPE by factors of 3 to 6 due to rapid chemical and deposition losses of HNO3 and other NOz species (e.g., Trainer et al., 2000). Additional discussion is found in Sect. 4.4.
In southern California, changes in the relative proportions of NOx-oxidation products have occurred and are thought to be instrumental in driving the rapid rates of O3 decline in that area (Pollack et al., 2013). These results indicate that measurements of HNO3 or PAN are needed to identify important changes in chemical pathways.
2.2 National O3 response to emission reductions
Between 1980 and 2013, the national average of the annual fourth-highest peak daily 8 h O3 mixing ratios, a metric relevant to the US O3 NAAQS, declined by 33 % (U.S. EPA, 2015d) as national VOC and NOx emissions decreased by 53 and 52 %, respectively (U.S. EPA, 2015e). Across the US and on multiple spatial scales from continental to urban, annual fourth-highest daily peak 8 h O3 mixing ratios between 1980 and 2013 show a statistically significant (p < 0.05) linear fit to either annual average or to 98th percentile daily maximum hourly NO2 mixing ratios; regression slopes are less than 1 : 1 and intercepts are in the range of 30 to 50 ppbv O3 (Hidy and Blanchard, 2015). Proportionalities between O3 and NO2 that are less than 1 : 1 are expected, and the observed intercept terms are approximately consistent with typical O3 mixing ratios of ∼ 20–50 ppbv observed at remote monitoring sites (Oltmans et al., 2008, 2013; U.S. EPA, 2012; Fiore et al., 2014; Lefohn et al., 2010, 2014; Cooper et al., 2012, 2014).
Although nonlinearity of O3 production and accumulation with respect to ambient VOC and NOx is well established (Lin et al., 1988), a tendency toward linearity is expected at sufficiently low NOx mixing ratios. As an example, the O3 photochemical production rate during June 1990 at Kinterbish, AL, was approximately linear over a range of ambient NOx from 0.1 to 2 ppbv (Trainer et al., 2000). Observed O3 extrema can also exhibit an apparent linear or near-linear response to ambient NOx mixing ratios if the extrema consistently fall within the lower-right quadrant (NOx-sensitive regime) of an O3–VOC–NOx diagram, a concise graphical representation first established empirically from southern California data and later generated using the Empirical Kinetic Modeling Approach (EKMA) (illustrated in Hidy and Blanchard, 2015). The O3–VOC–NOx diagram has been adopted by many investigators for displaying the output of box models (e.g., Fujita et al., 2003, 2015) and grid-based photochemical models (e.g., Reynolds et al., 2003, 2004).
Southern California historically has exhibited the highest peak O3 mixing ratios in the US since the 1960s. Because of high ambient O3 and precursor mixing ratios there and the complexity of the relationships of O3 with NOx and VOC, some investigators have described southern California O3 and precursor trends in terms of percentage changes. For example, Pollack et al. (2013) report that peak 8 h O3 mixing ratios in southern California declined exponentially over time at a rate of 2.8 % per year between 1973 and 2010, thus decreasing O3 levels by approximately a factor of 3. This rate of O3 decline exceeds rates occurring in other metropolitan areas (Hidy and Blanchard, 2015). O3 extrema in southern California decreased along with declining mixing ratios of ambient VOCs and NOx (7.3 % yr−1 and 2.6 % yr−1, respectively, 1960–2010) and declining ratios of VOC ∕ NOx (4.8 % yr−1) (Pollack et al., 2013). The rates of atmospheric oxidation of NOx increased over time and changes in NOx oxidation reactions increasingly favored production of HNO3, a NOx reaction product associated with radical termination and quenching of the O3 formation cycle (Pollack et al., 2013). To our knowledge, changes in the relative proportions of atmospheric reaction products accounting for rapid rates of O3 reduction have not been reported for locations other than southern California.
3.1 Emissions and ambient air quality measurements
Air quality monitoring data were obtained from the EPA Air Quality System (AQS) data archives for all sites in Georgia, Alabama, and Mississippi (U.S. EPA, 2016a). Daily measurement values (i.e., peak daily 8 h O3 mixing ratio) as well as annual summary statistics (e.g., maxima, annual averages) were acquired. We obtained deposition data from the two EPA Clean Air Status and Trends Network (CASTNET) monitoring sites located within the study region: Sand Mountain, AL (125 km east-northeast of the SEARCH site at Centreville), and Georgia Station, GA (102 km SE of the SEARCH site at Yorkville) (U.S. EPA, 2016b).
Annual, state-level emission trends data were obtained from the U.S. EPA (2016c, d), Xing et al. (2013), and Hidy et al. (2014). The comparability of inventories is discussed in the Supplement (Fig. S1). Because the EPA trend inventory utilized different methods for estimating mobile-source emissions prior to 2002 compared with 2002 and later years, we combined EPA trend estimates for 2002–2016 with the 1996–2001 emission estimates of Hidy et al. (2014), which are consistent with more recent EPA methods (see Supplement).
Hourly measurements of gases (NO, NO2, NOy, HNO3, and O3) were obtained from SEARCH public archives (ARA, 2017). All parameters measured at the sites are calibrated and audited to conventional reference standards, as described in ARA (2015). Network operations, sampling, and measurement methods are documented in Hansen et al. (2003, 2006); see also Table S1 in the Supplement. The network consisted of eight extensively instrumented monitoring sites located in the southeastern US along the Gulf of Mexico and inland (Fig. S2): Pensacola, Florida (PNS), and Gulfport, Mississippi (GFP), urban coastal sites (∼ 5 and 1.5 km from the shoreline, respectively); Pensacola – outlying (aircraft) landing field (OLF) and Oak Grove, Mississippi (OAK), non-urban coastal sites near the Gulf (∼ 20 and 80 km inland, respectively); Atlanta, Georgia – Jefferson Street (JST) and North Birmingham, Alabama (BHM), urban inland sites; and Yorkville, Georgia (YRK), and Centreville, Alabama (CTR), non-urban inland sites. PNS, OAK, and GFP were closed at the end of 2009, 2010, and 2012, respectively. SEARCH site locations are described in detail, including discussion of possible emission influences, in Hansen et al. (2003) and Hidy et al. (2014). SEARCH VOC data are available for JST as daily data from 1999 through 2008, and U.S. EPA VOC measurements are available for YRK as summer hourly data and as 24 h samples collected every sixth day throughout the year (Blanchard et al., 2010a). EPA VOC samples are also available for three other sites in the Atlanta area; only one of these additional sites reported data through 2014.
SEARCH meteorological parameters and gases are sampled at a height of 10 m, characteristic of lower-tropospheric mixing ratios near the surface (Hansen et al., 2003, 2006; Edgerton et al., 2007; Saylor et al., 2010). Gas and meteorological measurements commenced in 1992 at the rural sites of CTR, OAK, and YRK. The measurements at rural SEARCH sites included O3, NO, and NOy beginning in 1992, and NO2 and HNO3 measurements began in 1996. Consistent measurement methods have been utilized for all gases except NO2. NO2 measurements commenced network-wide in 2002, and three NO2 measurement methods have been employed during the network operations (Table S3). All three methods are NO2-specific, differing primarily in the light source used for photolysis of NO2. The NO2 data exhibit consistency with NO and NOy measurements but with some variations occurring during specific years (e.g., 2001 and 2002, Fig. S3). Because changes in NO2 measurement methods could affect the computed NOz (NOy–NO–NO2), we repeat some data analyses using HNO3 in place of NOz. As noted, HNO3 data also provide useful insight into NO2 termination reactions. HNO3 measurements are the difference between NOy and denuded NOy (Table S1; Hansen et al., 2006). The SEARCH measurements of NOy were designed to capture particulate nitrate and organic nitrates, as well as NO, NO2, HNO3, and other oxidized nitrogen species. The NOy sampler derives from the instrument identified in Williams et al. (1998) as Environmental Science and Engineering (ESE), which was one of five instruments for which measurements of NOy reproduced the sum of separately measured NOy species. Additional testing in 2013 showed that SEARCH NOy measurements agreed with the sum of measured mixing ratios of NO, NO2, HNO3, particulate nitrate, alkyl nitrates, and peroxy–alkyl nitrates (Hidy et al., 2014).
Trace gas calibrations were done daily for O3 and every third day for other gases. Reported detection limits (Table S1) are 0.05–0.1 ppbv for oxidized nitrogen species and 1 ppbv for O3 (Hansen et al., 2003, 2006). NO2 measurement uncertainties are estimated as ∼ 30 % prior to 2002 and ∼ 10 % after 2002 (Hansen et al., 2006). Measurement uncertainties are estimated to be 10 % or less for other oxidized nitrogen species and 5 % or less for ozone (2σ in all cases). Propagation of errors indicates corresponding 2σ measurement uncertainties averaging 0.5 ppbv for mid-afternoon NOz (< 0.1 ppbv for NOz < 1 ppbv) and 0.16 for the ratio NOz ∕ NOy.
3.2 Data analysis
Multiple methods were employed to characterize the variability of ambient O3 and NOy mixing ratios. Analyses of seasonal variability used data from all months of each year. Diurnal hourly average mixing ratios were computed by year to characterize patterns of temporal change and to identify hours associated with O3 maxima. Observed slopes of regressions of O3 versus NOz were computed as previously done in measurement studies using afternoon O3 and NOz data (Trainer et al., 1993, 1995; Kleinman et al., 1994; Hirsch et al., 1996; Kasibhatla et al., 1998; Nunnermacker et al., 1998; St. John et al., 1998; Sillman et al., 1998; Zaveri et al., 2003; Griffin et al., 2004; Travis et al., 2016). Because past studies have examined O3 formation in photochemically aged air (i.e., at locations distant from fresh emissions, where atmospheric reactions have acted on emissions from earlier times) during summers (e.g., Trainer et al., 1993), our analyses focus on the months of June and July to select weeks nearest maximum solar radiation (∼ −20, +40 days). Additional analyses were carried out for other months to facilitate comparisons across seasons. As for earlier studies, the calculations are based on afternoon times, using hourly values starting at 14:00 local standard time to represent the daily peak O3 after morning production and before mixing ratios decline with decreasing photochemical reaction in later afternoon. In addition to characterizing O3 ∕ NOz and its change with time, corresponding supporting analyses are presented for O3 ∕ HNO3. As a supplemental analysis, rates of maximum diurnal increase of O3 and HNO3 during late morning and early afternoon were computed for comparison of ΔO3 with ΔHNO3.
4.1 Trends
Hidy et al. (2014) report a 63 % reduction of NOx emissions in the southeastern US between 1996 and 2014. The largest NOx emission changes in the southeast occurred between 2007 and 2009 due to reductions of emissions from electric generating units (EGUs) and from diesel engine vehicles, and they were accompanied by more gradual year-to-year reductions of gasoline-engine mobile-source NOx emissions (de Gouw et al., 2014; Hidy et al., 2014). NOx emission reductions led to approximately proportional responses of mean ambient NOy and NOz mixing ratios at SEARCH sites (Hidy et al., 2014).
The EPA CASTNET data show wet and dry nitrate deposition since the late 1990s declining at rates of ∼ 5 % per year (−0.045 ± 0.005 and −0.056 ± 0.005 yr−1), nearly identical to NOx emission changes of −0.046 ± 0.001 and −0.051 ± 0.003 yr−1 (Fig. 1). Total (wet plus dry) nitrate deposition fluxes decreased linearly in proportion to reductions of NOx emissions in Alabama and Georgia (Fig. 1). Linear regression slopes indicate that the annual nitrate deposition fluxes at the Georgia and Alabama CASTNET sites correspond to 30 % of Georgia emissions and 36 % of Alabama emissions on an annual and statewide basis (Fig. 1). Emissions are not spatially homogeneous and deposition losses likely vary with distance from emission sources. The two sites are situated differently in relation to metropolitan areas, possibly affecting deposition fluxes; Sand Mountain (SND) is northeast of Birmingham and Georgia Station (GAS) is south of Atlanta. The linearity and statistical significance of the regressions indicate that the fraction of NOx emissions lost to deposition has not changed over time (ratios of annual deposition to state emissions varied without trend from 0.23–0.34 at GAS and 0.30–0.45 at SND). Mean annual SEARCH NOy mixing ratios at rural CTR and YRK declined at ∼ 5–7 % yr−1 (Fig. S4). SEARCH and EPA CASTNET sites exhibit downward trends in mean annual HNO3 concentrations of ∼ 9–11 and ∼ 6–7 % yr−1, respectively (Fig. S4). Ambient NOy and HNO3 trends are not statistically different from state-level NOx emission trends.
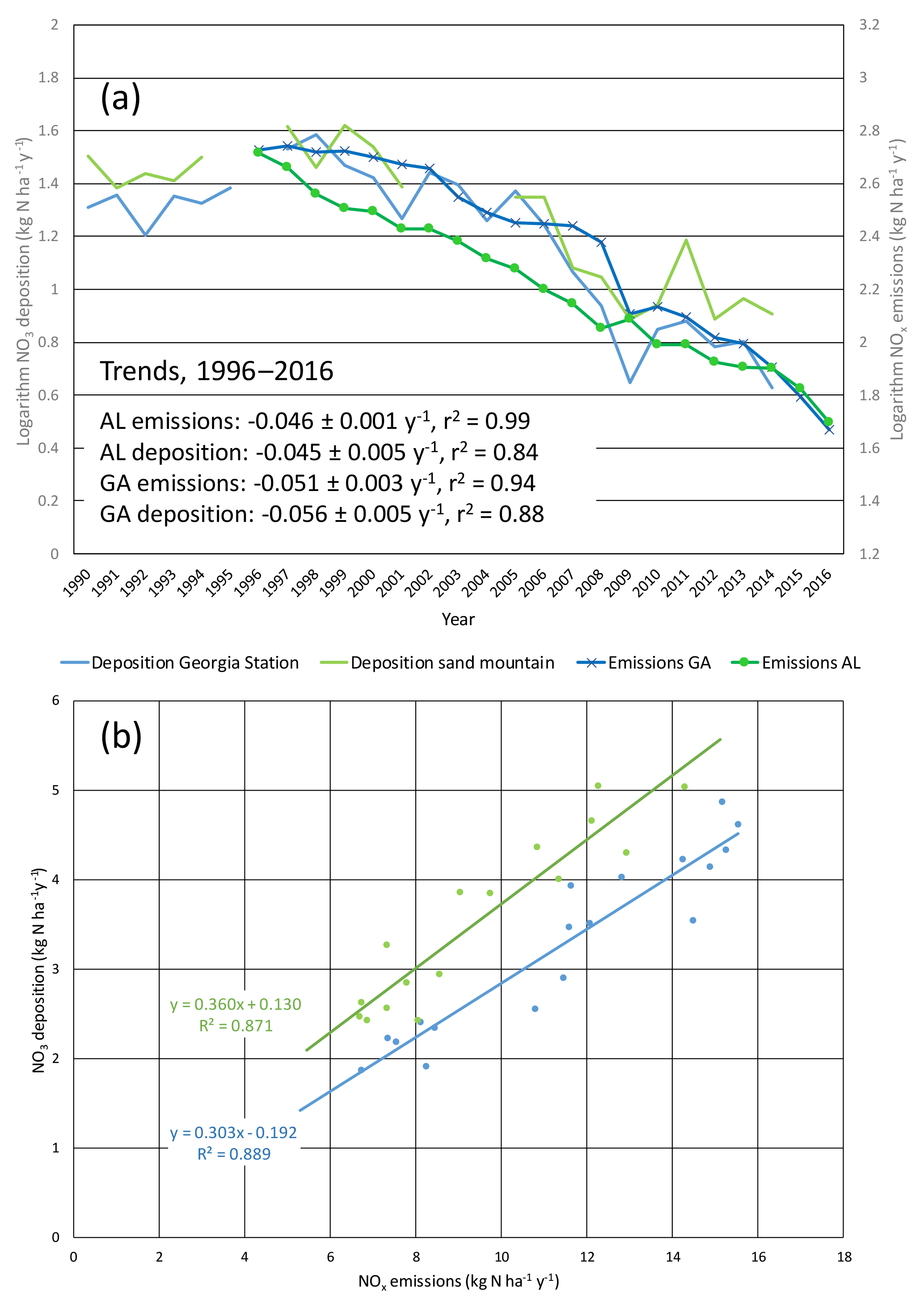
Figure 1Comparison of nitrate deposition (wet plus dry) to NOx emission densities in Georgia and Alabama as (a) temporal trends and (b) regression of deposition against emissions (with same color coding in both panels). Nitrate deposition and NOx emission densities are expressed as kg ha−1 yr−1. NOx emissions are from all source sectors (see Supplement). Panel (a) shows natural logarithms vs. year and indicates that emissions and deposition trended downward at the same rates. Panel (b) slopes are statistically significant (p < 0.0001) and intercepts are not (p > 0.1).
Annual fourth-highest daily peak 8 h O3 mixing ratios at compliance monitoring sites in Georgia, Alabama, and Mississippi exhibit statistically significant (p < 0.0001) linear correlations with annual NOx emissions in those states between 1996 and 2015 (Fig. 2), qualitatively consistent with past work indicating that high O3 would respond to reductions of NOx emissions (Chameides and Cowling, 1995; Jacob et al., 1995; Kasibhatla et al., 1998). Intersite differences in the annual fourth-highest daily peak 8 h O3 mixing ratios have decreased (Fig. 2), consistent with an analysis of data from a larger number of US and European locations (Paoletti et al., 2014). The annual fourth-highest daily peak 8 h O3 mixing ratios are declining toward nonzero values, as indicated by the statistically significant (p < 0.0001) intercepts of ∼ 45–50 ppbv (Fig. 2). SEARCH data are used to characterize the southeastern O3 response to emission changes in greater detail. Between 1999 and 2014, the highest peak daily 8 h O3 mixing ratios occurring each month (monthly O3 maxima) declined at all SEARCH sites at statistically significant (p < 0.01) rates averaging ∼ 1–1.5 ppbv yr−1 (Fig. 3). These declines are comparable to the trend in the 95th percentile summer peak daily 8 h O3 mixing ratios in the southeastern US of ∼ −0.8 to −1.8 ppbv yr−1 reported by Lin et al. (2017), with downward trends occurring in other seasons as well. The observed SEARCH O3 trends are also consistent with other analyses of North American observations (e.g., Chan, 2009; Lefohn et al., 2010; Paoletti, 2014; Simon et al., 2015) and with the trends occurring at EPA monitors in the southeast (Fig. 2). Both EPA (Fig. 2) and SEARCH (Fig. 3) data suggest that O3 mixing ratios increased during the 1990s, then began declining. This result is consistent with modeling by Reynolds et al. (2004), which predicted an initially slow response of Atlanta-area O3 to emission reductions between 1996 and 2000, followed by increasing sensitivity to reductions of NOx emissions. The observed O3 decreases exceed those predicted by Reynolds et al. (2004) because actual NOx emission reductions (∼ 60 % between 1996 and 2014) exceeded the projected emission reductions (∼ 40 % by 2010 and ∼ 55 % by 2020) that were anticipated based on known and anticipated emission control rules at the time of the modeling study. The SEARCH trends are compared with emission changes in the southeast and with emission and O3 trends in southern California, in Table S2.
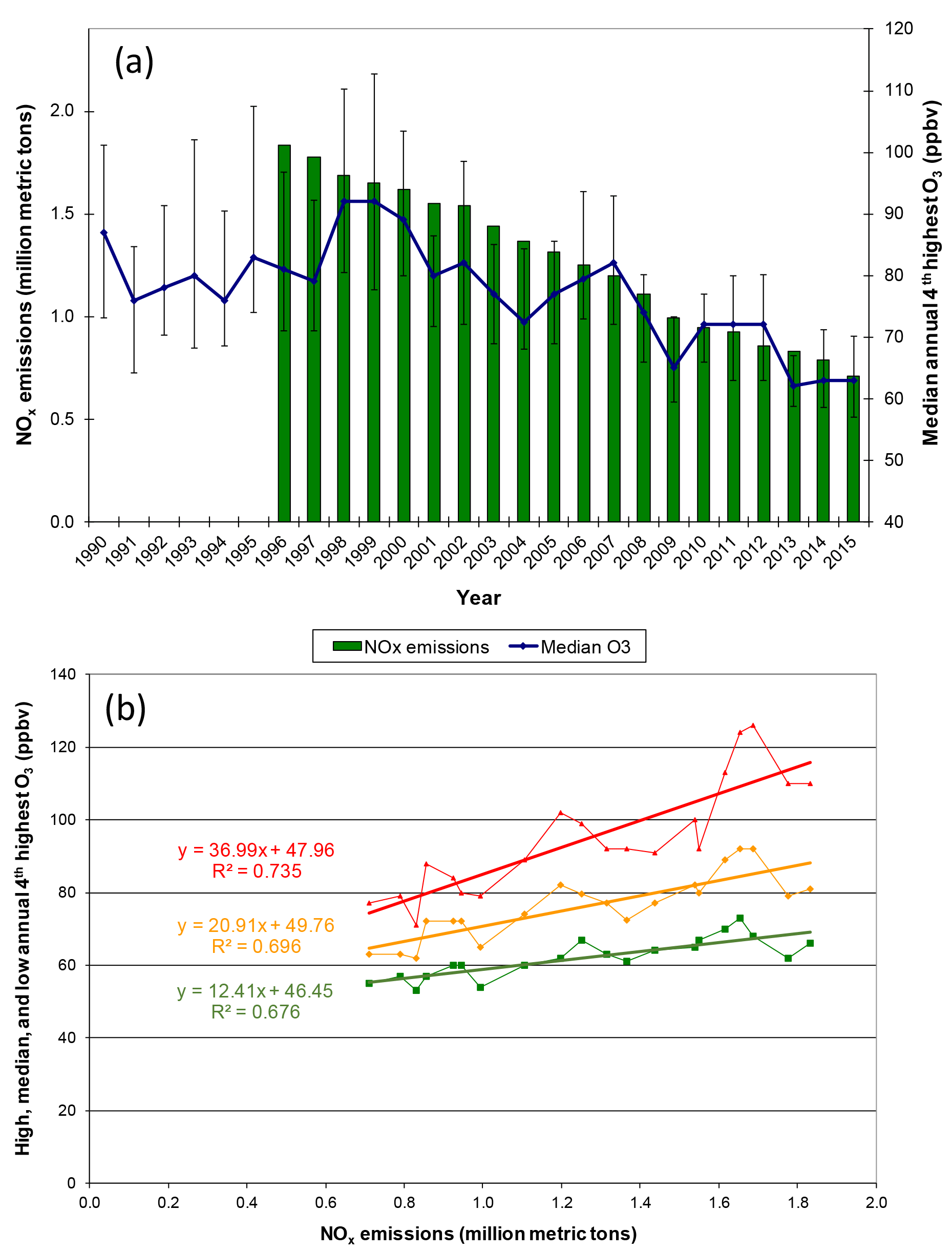
Figure 2Comparison of annual fourth-highest daily peak 8 h O3 to NOx emissions in Georgia and Alabama (a) trends (±90th and 10th percentile sites) and (b) regressions (high = 90th percentile site, median, and low = 10th percentile site annual fourth-highest daily peak 8 h O3). NOx emissions are from all source sectors (see Supplement). O3 data include all EPA AQS monitors in Georgia and Alabama for each year having at least 75 % data completeness (mean = 55 monitors, low of 32–36 in 1990–1993). Slopes and intercepts are statistically significant (p < 0.0001).
More complete understanding of regional O3 trends requires consideration of both regional emission changes and possible changes in background O3. Multiple definitions of the term “background O3” may be found in the literature, including global background, continental background, non-anthropogenic background, and regional background, among others. For the O3 trends shown in Figs. 2 and 3, the most relevant consideration is the amount of O3 transported into the study domain across upwind boundaries (denoted here as regional background or transported O3). The percentage reductions of O3 are larger if transported O3 can be estimated and subtracted from observed O3 mixing ratios, and this adjustment potentially provides a better assessment of the effects of regional emission reductions on the fraction of O3 that is manageable by means of local and regional emission control measures. For example, Parrish et al. (2017a) report that the O3 enhancement above background in southern California decreased by 4.5 % yr−1, which is larger than the unadjusted O3 decline of 2.8 % yr−1 given by Pollack et al. (2013). Similarly, rates of decline in southeastern US O3 are larger if regional background O3 is considered (Table S2).
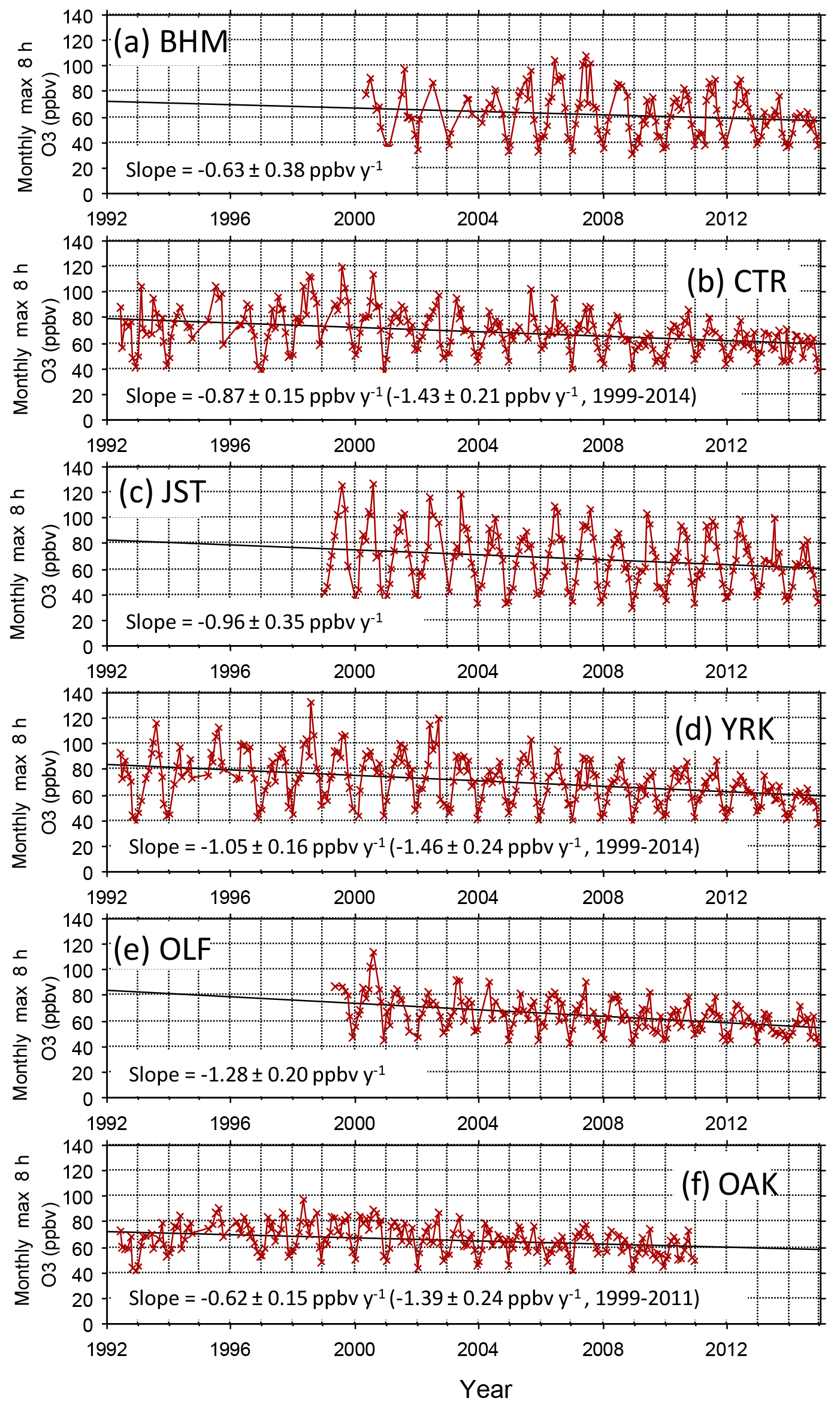
Figure 3Monthly maxima of daily peak 8 h average O3 mixing ratios. All monthly maxima are determined from 24 or more days with 18 or more sampling hours per day. PNS and GFP (not shown) exhibit trends of −1.64 ± 0.45 and −0.60 ± 0.32 ppbv yr−1, respectively. Trends are statistically significant (p < 0.01) at CTR, JST, OAK, OLF, PNS, and YRK.

Figure 4Monthly means of daily peak 8 h average O3 mixing ratios. All monthly means are determined from 24 or more days with 18 or more sampling hours per day. Standard errors of the means average 2 (range 0.8–5) ppbv.
Defining and estimating regional background (or transported) O3 are each challenging. We interpret the intercepts in Fig. 2 as indicators of mean O3 levels that would occur on days with weather conducive to high O3 in the absence of NOx emissions from AL and GA sources, i.e., as estimators of O3 transported into the region from outside the study domain (as discussed subsequently, multi-day carryover of local and regional emissions during stagnation events could also affect intercepts and slopes). Days with weather that is not conducive to high O3 likely have different levels of transported O3. The statistically significant slopes in Fig. 2 indicate O3 enhancements that are attributable to AL–GA emissions, except as noted next, and a comparison of the O3 decline to intercept-corrected O3 would then reveal the proportionality between AL–GA emissions and AL–GA O3 enhancements over O3 originating outside the study domain (i.e., in excess of regional background O3). Although the ∼ 30–35 % O3 declines are less than proportional to the ∼ 60 % decrease in NOx emissions, the decline in the median O3 is ∼ 60 % if the 50 ppbv intercept shown in Fig. 2 is subtracted from the O3 mixing ratios.
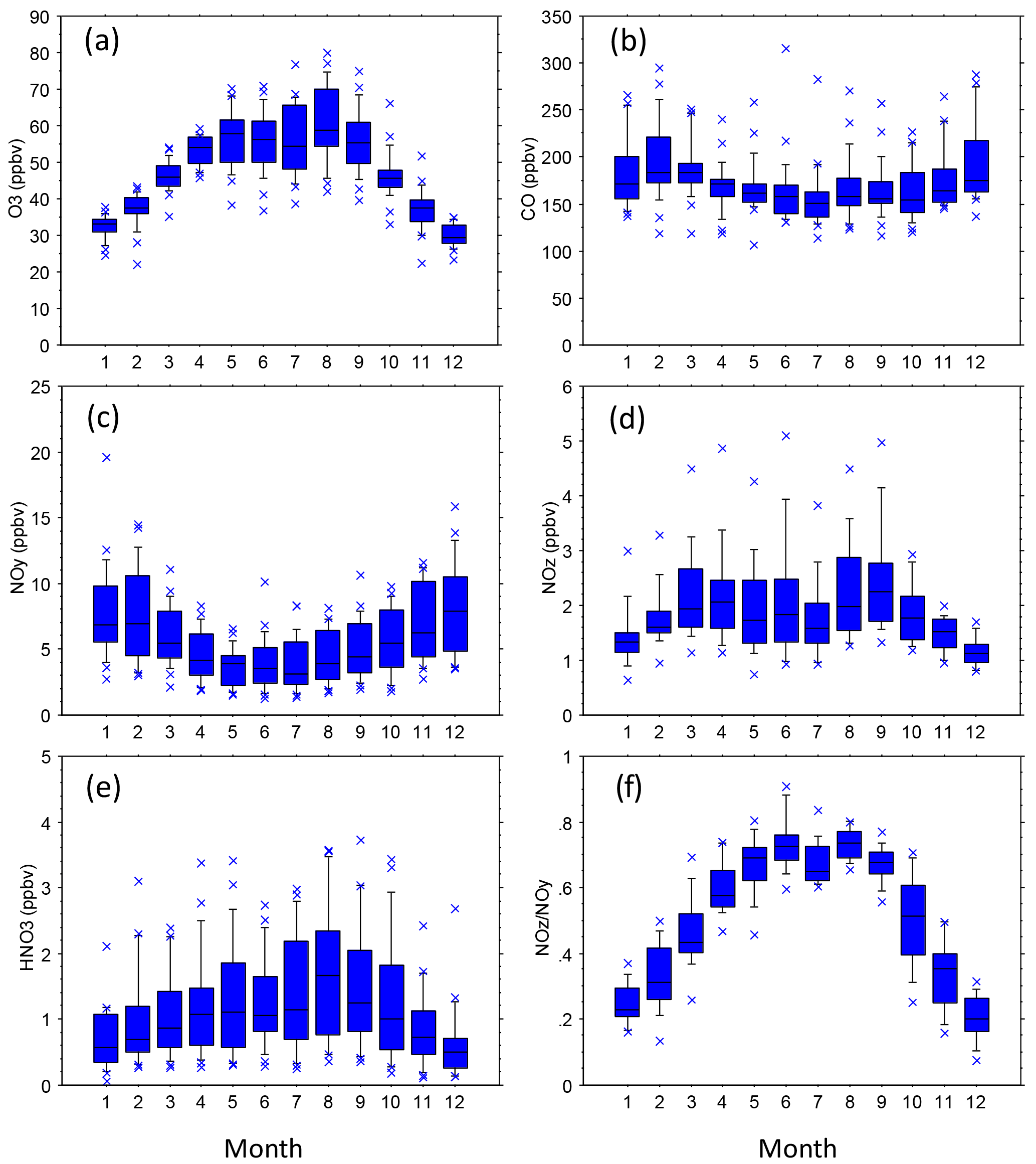
Figure 5Statistical distributions of mean monthly species mixing ratios (all SEARCH sites, 1992–2014). Distributions indicate the 10th, 25th, 50th, 75th, and 90th percentiles of the monthly averages.
If the amount of O3 that has been transported from upwind regions has been changing over time, e.g., declining as NOx emissions and ambient O3 decline in adjacent regions, the slopes shown in Fig. 2 would reflect changes in both the O3 that originated upwind and in the O3 enhancements attributable to AL–GA emissions, confounding attribution. Related studies do not provide consistent evidence for a trend, either upward or downward, in regional background O3 in the southeastern US. For example, baseline O3 concentrations in air flowing into Texas from the Gulf of Mexico during May through October did not change significantly between 1998 and 2012 (Berlin et al., 2013). Mean regional background O3 mixing ratios were 48 to 59 ppbv in the Houston, TX, area on days with O3 levels exceeding the NAAQS, which includes O3 contributions from transport to the area from other regions of the US (Berlin et al., 2013). Observed trends in the 5th percentile O3 have previously been used as indicators of changes in either regional or continental background O3 (e.g., Wilson et al., 2012). The 5th percentile peak daily 8 h O3 mixing ratios decreased during summer at rural sites throughout the southeastern US between 1988 and 2014 (Lin et al., 2017). By this measure, regional background O3 levels did not increase in the southeastern US during our study period.
Large-scale transport affecting O3 in the boundary layer and at the surface is a function of altitude. For example, during June 2013, anthropogenic emissions and long-range transport (long-range tropospheric + stratospheric) O3 each accounted for about 40 % (15–20 ppbv each) of model-predicted O3 below 1 km altitude at Huntsville, AL, while long-range transport accounted for ∼ 80 % of model-predicted O3 above 4 km altitude (Johnson et al., 2016). This variation of source contributions with altitude provides an opportunity to differentiate between emission-related and transport-related trends derived from vertical soundings of upper-air O3 mixing ratios. Using ozonesondes that are generally launched on a weekly schedule, vertical O3 mixing ratio profiles have been determined by the University of Alabama in Huntsville, Alabama, since 1999 (Newchurch et al., 2003; Johnson et al., 2016; University of Alabama Huntsville, 2017; NOAA, 2017). We obtained these ozonesonde data (n=940 days) and identified the following statistically significant trends in the lower layers that are relatively more influenced by local and regional emissions according to Johnson et al. (2016): −0.25 ± 0.11 ppbv yr−1 (p < 0.05) in daily measurements at 0.5 km, −0.40 ± 0.10 ppbv yr−1 (p < 0.0001) at 1 km (daily), −0.42 ± 0.09 ppbv yr−1 (p < 0.0001) at 2 km (daily), and −0.57 ± 0.13 ppbv yr−1 in monthly averages of O3 measurements made throughout the interval 1–2 km (p < 0.001). At higher altitudes where Johnson et al. (2016) predicted that long-range transport is the dominant source of O3, no trends occurred: 0.06 ± 0.08 ppbv yr−1 (p > 0.1) at 4 km (daily) and 0.09 ± 0.19 ppbv yr−1 (p > 0.1) at 8 km (daily).
Global background is one component of regional background and trends in global background are expected to contribute to trends in regional background. Lin et al. (2017) show that rising NOx emissions in Asia have increased modeled North American background O3 levels (based on model simulations with zero North American emissions) by ∼ 0.2 ppbv yr−1 in the southeastern US in summer, which is a small effect even when accumulated over 20 years in comparison with the ∼ 25 ppbv reduction in the median annual fourth-highest peak daily 8 h O3 shown in Fig. 2. Multiple studies have demonstrated increasing trends in global background O3 mixing ratios (Ordóñez et al., 2007; Oltmans et al., 2008; Arif and Abdullah, 2011; Wilson et al., 2012). Parrish et al. (2017a) report that the highest O3 design values (the 3-year running mean of the annual fourth-highest peak daily 8 h O3 mixing ratio) in southern California are converging toward a limit of 62.0 ± 1.9 ppb, which they identify as the O3 design values that would result from US background O3 concentrations. Parrish et al. (2017b) report decreasing O3 transported across the Pacific into the western US after 2000. As noted, regional background O3 in the southeastern US does not appear to be trending either upward or downward, even though trends in background O3 have been established in other areas or globally.
In the southeastern US, the simple conceptual model of O3 transported into a study region across upwind boundaries is incomplete. High O3 typically occurs during multi-day stagnation episodes, which are associated with the presence of high barometric pressure over the domain and limited transport (Blanchard et al., 2013). Transport distances determined from 24 h back-trajectory computations are less than 300 km for the highest decile O3 (Blanchard et al., 2013). Mean 24 h transport distances are less than 350 km during June and less than 380 km during July (Blanchard et al., 2014). These distances are approximately equivalent to distances from Birmingham to Mobile, AL, or from Atlanta to Savannah, GA. Local and regional emissions can accumulate over multiple days and potentially could contribute to observed O3 concentrations (e.g., aloft) that are considered as regional background. In contrast to emissions originating upwind, carryover from emission sources within the study domain is a manageable component of efforts to reduce O3.
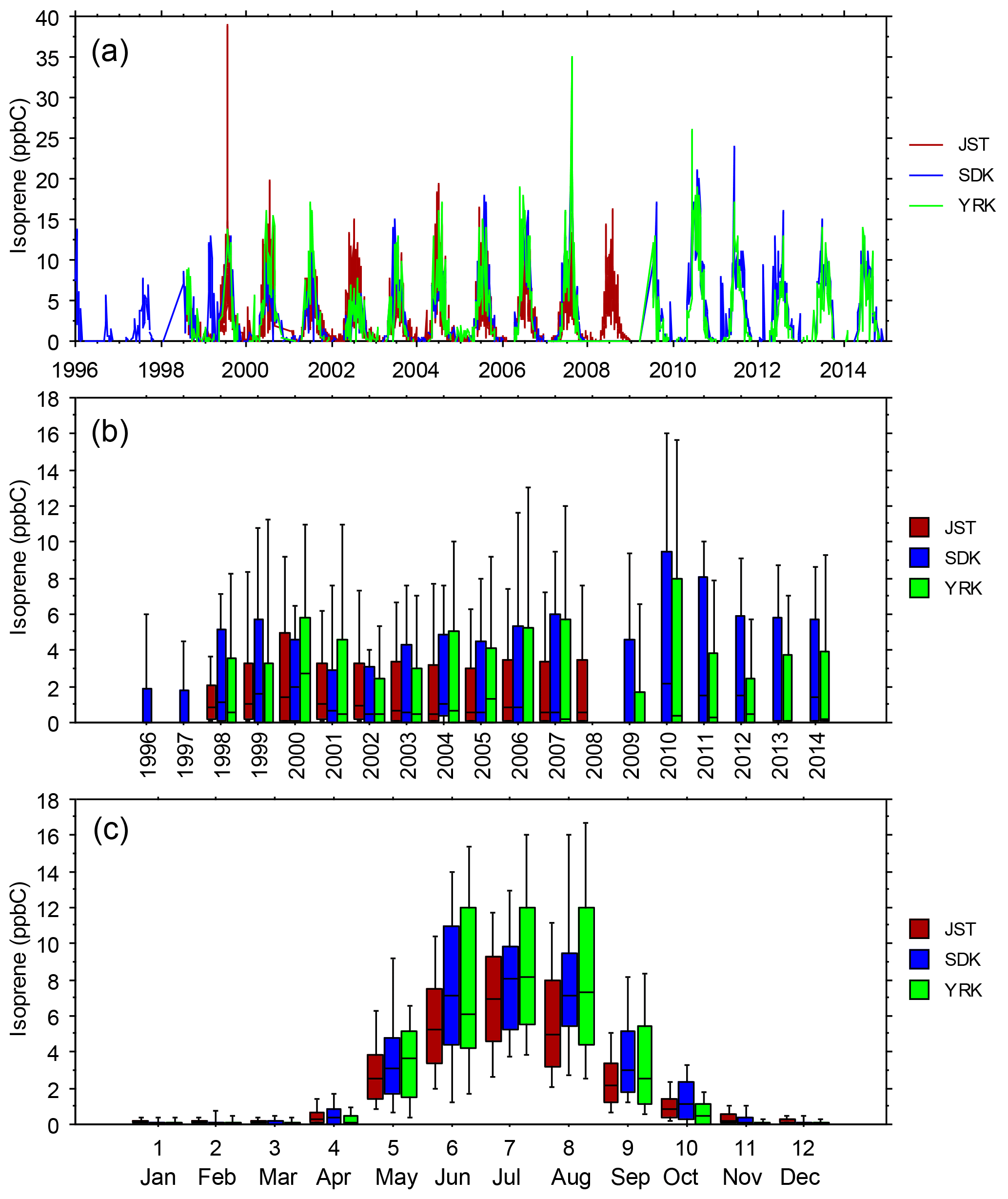
Figure 6(a) Daily-average isoprene mixing ratios vs. date, (b) statistical distributions of daily-average isoprene mixing ratios vs. year, and (c) statistical distributions of daily-average isoprene mixing ratios vs. month. Samples were obtained every day at JST and once every 6 days at YRK and SDK (Blanchard et al., 2010a). Distributions indicate the 10th, 25th, 50th, 75th, and 90th percentiles.
4.2 Seasonal variations of O3, NOy, NOz, HNO3, and VOCs
The seasonal oscillations of monthly O3 maxima in the southeast are coupled to local or regional meteorology, solar radiation, and emissions (e.g., Blanchard et al., 2013, 2014; Hidy et al., 2014). Variations of daily maximum temperature and midday relative humidity (RH) are associated with variations of daily peak 8 h O3 mixing ratios by ∼ ±30 % from mean peak 8 h O3 mixing ratios, after also accounting for variations of other meteorological factors (Blanchard et al., 2014). Air mass back trajectories originating from the south (∼ 150 to 200∘) exhibit peak 8 h O3 that is ∼ 5–10 % lower than average; daily peak O3 decreases as 24 h back-trajectory distances increase from zero to ∼ 600 km, consistent with association of higher O3 concentrations with air mass stagnation rather than transport (Blanchard et al., 2013, 2014). At SEARCH sites, the monthly O3 maxima (highest daily peak 8 h O3 each month) and mean daily peak 8 h O3 mixing ratios typically occurred in summer months, especially inland, and declined more than other monthly maxima (Figs. 3 and 4). Summer means were not always higher than spring averages, especially at rural and coastal sites and during more recent years (Fig. 4). Roughly constant winter monthly peak 8 h maxima of ∼ 40 ppbv occurred throughout the period of record (Fig. 3). The seasonal variability of the highest peak daily 8 h O3 therefore declined over time (see also Table S3). Similar results were found for monthly means of hourly measurements, discussed in Sect. 4.3 on diurnal variations. Other recent studies have reported decreasing seasonal variability of O3 across the US using data from large numbers of monitoring sites (Chan, 2009; Chan and Vet, 2010; Cooper et al., 2012; Paoletti et al., 2014; Simon et al., 2015). Declines in seasonal variability are thought to result from changing rates of O3 formation as precursor emissions have declined or from increasing influence of intercontinental background O3, not from changes in seasonal variations of temperature and other meteorological factors (Chan, 2009; Cooper et al., 2012; Simon et al., 2015).

Figure 7Average O3 mixing ratios vs. hour, by year. Each data point is the mean of all hourly measurements during June through August. Sites at PNS and GFP (not shown) exhibit similar diurnal profiles and trends (sampling at those sites ended after 2009 and 2012, respectively). Standard errors of the means are 0.3–4 ppbv (∼ 2 % of mean O3 mixing ratios).
The SEARCH data indicate that seasonal variations occur in ambient O3, NOy, NOz, HNO3, and the ratio of NOz ∕ NOy (Fig. 5). Seasonal variations of temperature and other meteorological factors are known to cause seasonal variations of O3 and NOy species concentrations. The monthly average NOz and HNO3 mixing ratios indicate that active photochemical processing of NOx occurs during well over half the year in the warm climate of the southeastern US. The effects of VOC species on O3 formation depend on both their ambient concentrations and their reactivities. To describe VOC variations at sites with long-term VOC measurements, we use isoprene data as an indicator of biogenic VOCs and toluene as an indicator of anthropogenic VOCs (nominally emitted as a gasoline vapor). The importance of isoprene emissions for O3 production in the southeastern US is well established (e.g., Chameides et al., 1988; Chameides and Cowling, 1995; Frost et al., 1998; Starn et al., 1998; Wiedinmyer et al., 2006; Zhang et al., 2014; Lin et al., 2017). We also consider other reactive VOC species of interest, including α-pinene (biogenic) as well as ethylene and xylenes (anthropogenic). Summer (June–August) months exhibit elevated ambient mixing ratios of rural and urban isoprene, typically about 5–10 ppbC, that are 1 to 2 orders of magnitude greater than those occurring between October and April (Fig. 6). Transitions between low and high ambient isoprene mixing ratios occur in mid-May and mid-September in northern Georgia (Fig. 6). Annual mean isoprene mixing ratios were relatively constant, ∼ 2.5–3 ppbC, between 1998 and 2014. Biogenic VOCs, primarily isoprene, represent ∼ 20 % of the VOC reactivity at JST, ∼ 30 % at South DeKalb (SDK, located in metropolitan Atlanta ∼ 16 km southeast of JST), and ∼ 50 % at YRK, averaged over all samples collected between 1999 and 2007 (Blanchard et al., 2010a). Through precursor interactions, seasonal variations in isoprene mixing ratios are expected to affect seasonal variations in O3 mixing ratios and production rates.
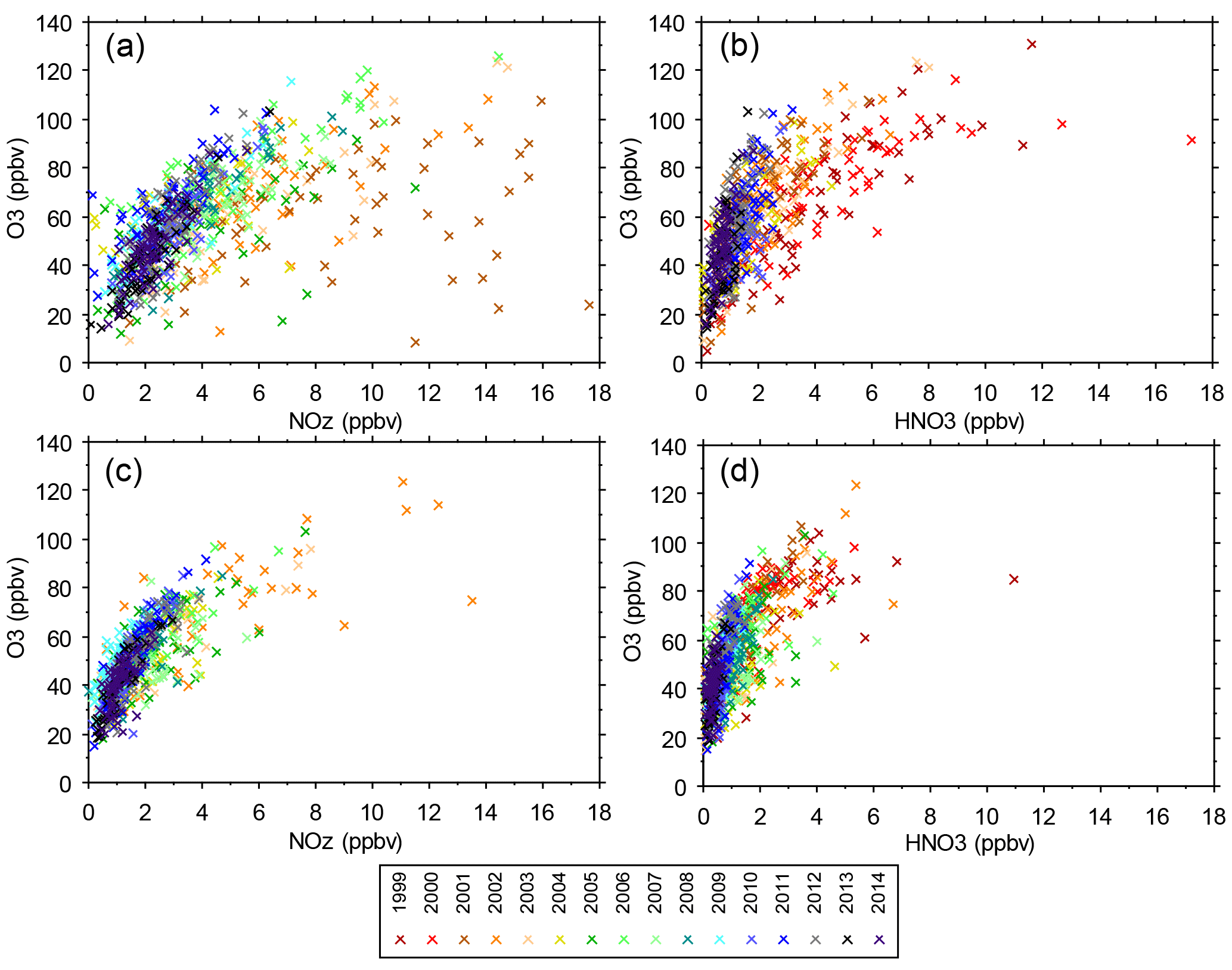
Figure 8(a) O3 vs. NOz at JST, (b) O3 vs. HNO3 at JST, (c) O3 vs. NOz at YRK, and (d) O3 vs. HNO3 at YRK. Each point is the 14:00–15:00 hourly average on 1 day, limited to days in June or July and delineated by year. The 2001 and 2002 NOz data may be biased high due to lower NO2 mixing ratios obtained by the instrumentation used at that time (Fig. S2).
Mean mixing ratios of ethylene and aromatic compounds vary substantially between urban and rural sites and exhibit less, and a different, seasonal variation than does isoprene, peaking in the fall rather than in the summer (compare Figs. 6, S5, S6). Daily-average mixing ratios of toluene, xylenes, and ethylene decline over the years, consistent with regulatory reductions of anthropogenic VOC emissions (Figs. S5, S6). Seasonal variations in ambient mixing ratios and trends in the anthropogenic emissions of aromatic compounds are expected to influence O3 mixing ratios and production in urban settings (rural anthropogenic VOC mixing ratios are lower but detectable).
The 24 h average VOC mixing ratios are of somewhat limited value for showing the influence of VOCs on O3 formation and accumulation. VOC influence is dependent on NOx mixing ratios, which vary depending on proximity to emission sources and time of day. Meteorological variability, including diurnal and day-to-day changes in temperature, vertical mixing, cloud cover, photolysis, and air mass transport, further obscures the quantitative effects of VOCs on seasonal and interannual variations of O3. Influences of anthropogenic VOCs at SEARCH sites have previously been reported (Blanchard et al., 2010b, 2014) and are not analyzed beyond this summary.
4.3 Diurnal variations of O3, NOy, NOz, and HNO3
Summer (June–August) mean O3 mixing ratios exhibit characteristic nocturnal minima and midday (noon to 16:00, midpoint ∼ 14:00) maxima at all SEARCH sites (Fig. 7). This diurnal pattern remained essentially the same at both the urban and rural sites from 1999 through 2014, but the daytime maxima decreased. Between 1999 and 2014, the summer mean midday maxima declined by ∼ 30 ppbv at all sites, while nocturnal means exhibited variable responses (Fig. 7). Similar diurnal variations occur throughout the year, with smaller decreases in the mean midday O3 maxima occurring during seasons other than summer (Figs. S7–S9). By the end of the study period, diurnal O3 profiles were higher during spring (March through May) than summer at the rural sites (CTR and YRK, Figs. S7 and S8), consistent with the reduction in summer mean monthly daily peak 8 h O3 averages (Fig. 4). Decreasing summer diurnal mean NOy, HNO3, and NOz mixing ratios were also observed, with a general flattening of the profiles and with the times of maxima remaining consistent (Figs. S10–S12). O3 changes are discussed in relation to changes in NOy and NOz in Sect. 4.4, with emphasis on summer and additional consideration of spring months.
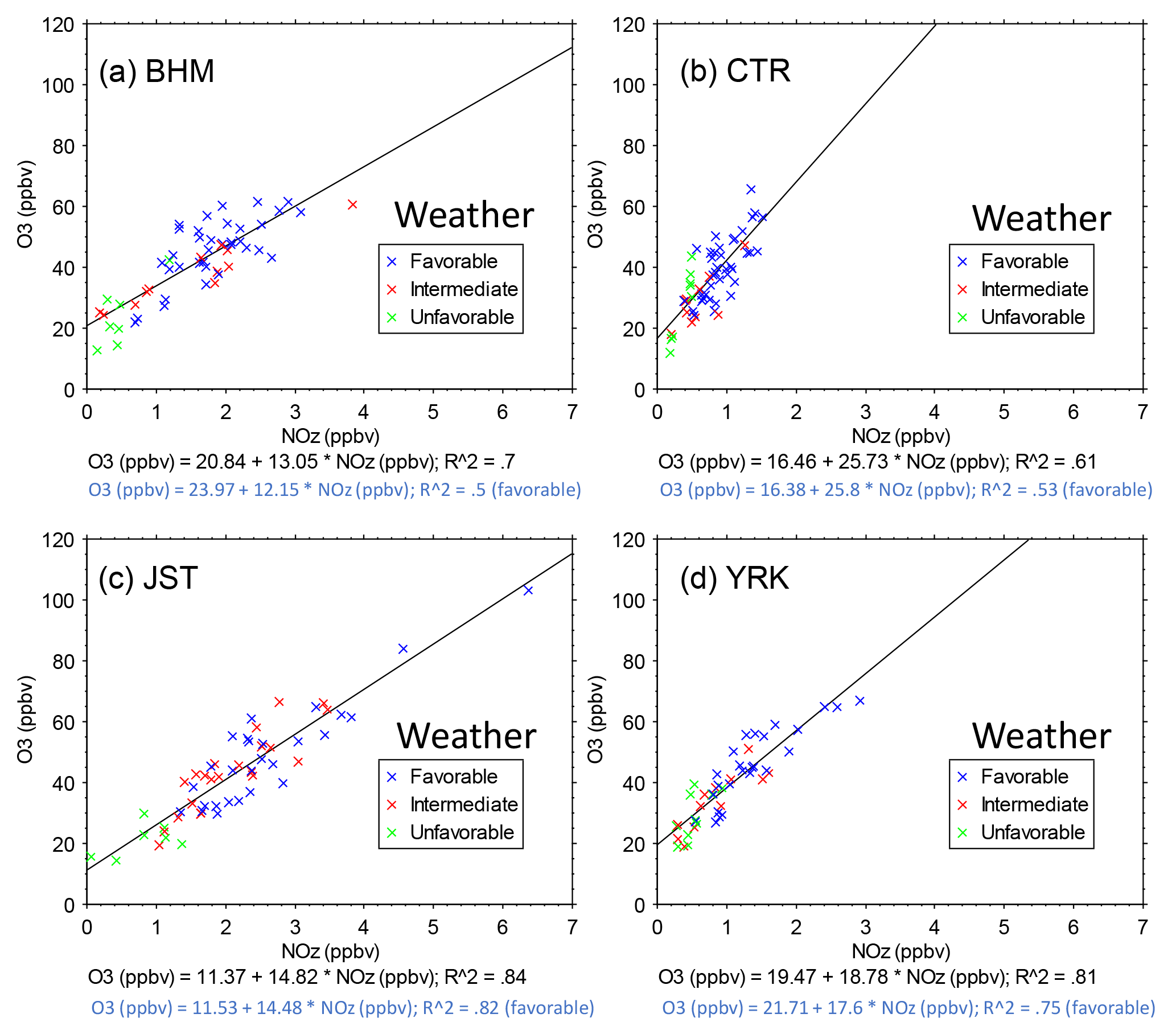
Figure 9O3 vs. NOz during June and July 2013. Each point is the 14:00–15:00 hourly average on 1 day. The data were selected to represent the approximate midpoint of the midday O3 maxima and to span a period around the summer solstice (∼ −20, ∼ +40 days) when solar radiation is highest on average. The regression slopes show higher rural than urban values: BHM = 13.05 ± 1.19 ppbv ppbv−1, JST = 14.82 ± 0.88 ppbv ppbv−1, YRK = 18.78 ± 1.38 ppbv ppbv−1, and CTR = 25.73 ± 2.76 ppbv ppbv−1. Corresponding regression slopes for Ox vs. NOz are BHM = 12.00 ± 1.16 ppbv ppbv−1, JST = 13.88,± 0.93 ppbv ppbv−1, YRK = 18.85 ± 1.37 ppbv ppbv−1, and CTR = 25.79 ± 2.79 ppbv ppbv−1. Symbols indicate the favorability of weather to O3 formation and accumulation: (1) favorable – T > 25 ∘C, RH < 70 %, and solar radiation > 500 W m−2; (2) intermediate – neither favorable nor unfavorable; (3) unfavorable – T < 25 ∘C, RH > 70 %, and solar radiation < 500 W m−2. Regression results are shown for all days and for the days with favorable weather.
4.4 Observed relationships between O3 and NOz
As discussed above, O3 mixing ratios vary seasonally and diurnally in response to variations in emissions, weather, background O3, and other factors. To reduce the influence of seasonal and diurnal variability, this section focuses on mixing ratios of NOz, HNO3, and O3 at 14:00 during June and July. Both temperature and solar radiation are typically high during June and July, and multi-day stagnation events occur frequently in association with high barometric pressure (Blanchard et al., 2013). Exceptions exist during the passage of frontal systems (Blanchard et al., 2013; Fig. S13). The 14:00 hour has the highest, or close to highest, average hourly O3 for all sites and years (Fig. 7). The atmosphere is well mixed by midday. Over the range of ambient mixing ratios observed across 15 years, the June–July 14:00 O3 values are distinctly nonlinear in relation to ambient NOz and HNO3 mixing ratios (Fig. 8). More variability is evident at urban sites than at rural sites, consistent with influence of urban NOx and perhaps VOC emissions on O3. The nonlinearity indicated in Fig. 8 is also evident when the data are restricted to days having the highest peak daily 8 h O3 mixing ratios (Fig. S14).
4.4.1 Linear models
Linear regressions are fit to the afternoon data by year, as shown in Fig. 9 for 2013 and in Table S4 for all years. During multi-week periods within any summer, all sites exhibit near-linear relationships of midday O3 to NOz. Because the ranges of NOx and NOz mixing ratios within each year are limited, year-specific relationships are close to linear and linear models are statistically significant. Steeper slopes at rural sites than at urban sites in Fig. 9 suggest that either more O3 molecules formed per molecule of NOx consumed in rural locales than in urban areas or that greater losses of NOz occurred at the rural sites, as discussed below. At all sites, similar results are obtained for regressions of Ox (O3 + NO2) vs. NOz compared with O3 vs. NOz (Fig. 9, caption). At 14:00, rural O3 mixing ratios are nearly identical with Ox mixing ratios and with other metrics (e.g., O3–[NOy–NO]) (Fig. S15). At urban sites, 14:00 NO2 mixing ratios are non-negligible, but this difference alters the intercepts rather than the slopes of the regressions of Ox vs. NOz compared with O3 vs. NOz (Fig. 9). As previously noted (Fig. S13), even during the 2-month periods that we analyzed, the weather is not always conducive to O3 formation and such days could influence the observed slopes and intercepts. However, regression results restricted to days with weather that favors O3 formation (as defined in Fig. 9) do not differ from the unrestricted regressions.
Plotting the year-specific (June–July) computed regression slopes versus mean June–July 14:00 NOz shows significant increases over time as ambient NOz mixing ratios have decreased, subject to year-to-year variability (Fig. 10, Table S4). Similar urban–rural differences and patterns of increasing regression slopes are also observed when data are restricted to March and April (spring) at YRK and JST (Fig. S16). The results for spring show more variability than the summer year-specific linear models. One key difference between spring and summer days is that cumulative solar radiation between sunrise and 14:00 is greater on summer days than on spring days, presumably fostering greater photochemical extent of reaction and accumulation of O3 during summer.
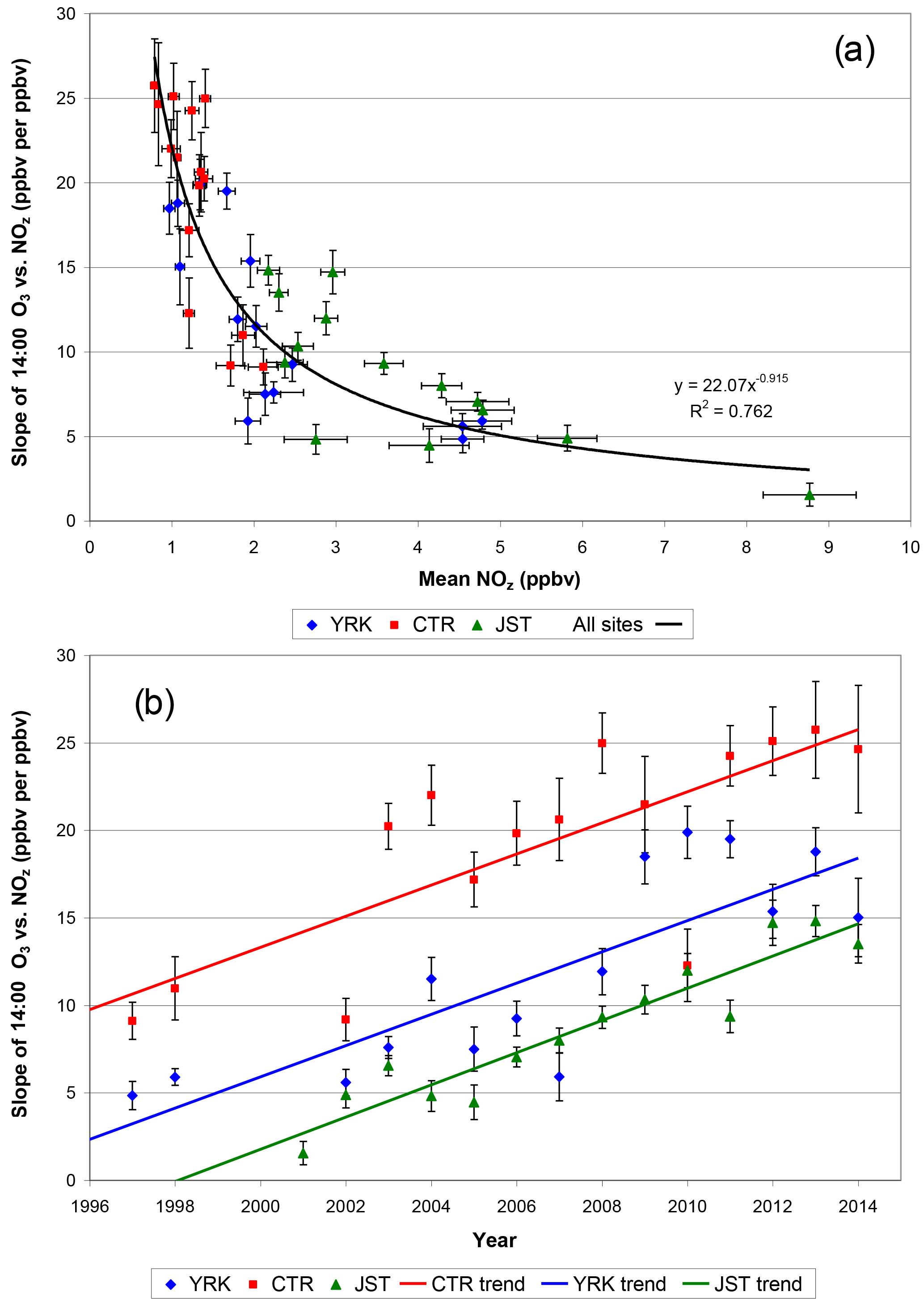
Figure 10(a) Summer CTR, JST, and YRK slope of daily (14:00) O3 and NOz vs. mean (14:00) NOz mixing ratios and (b) summer regression slope vs. year. NO2 data were not available for 1999 through 2001. Vertical and horizontal error bars are 1 standard error of the regression slopes and 1 standard error of the NOz means, respectively. Mean NOz measurement uncertainty is estimated as 0.2 ppbv (1σ).
The regression slopes determined from 14:00 data could reflect day-to-day differences in transported O3 if background O3 is consistently higher on high-O3 days than on low-O3 days and NOz is not (in contrast, random variations in day-to-day background O3 and NOz would introduce variations, or scatter, around the regression lines). We checked for an effect of this type by repeating the analyses using differences in mixing ratios. Two sets of difference-based regressions are used: (1) the differences between 14:00 and 10:00 hourly measurements and (2) the differences between 11:00 and 10:00 hourly measurements. The differences are computed for each day to minimize or eliminate the unknown day-specific background levels and are then used in the regressions. These hours were selected to focus on times of day when the atmosphere is well mixed. The morning rise in mixing heights is expected to contribute to increases in the mixing ratios of secondary species as aged air aloft is incorporated into the mixed layer. The most rapid rates of increase in diurnally averaged O3, NOz, and HNO3 values occur between ∼ 08:00 and 12:00 noon local time (Figs. 7, S8–S9). By mid- to late-morning hours during summer, considerable vertical entrainment has occurred, and subsequent changes in the mixing ratios of secondary species likely reflect same-day atmospheric chemical reactions. Computing afternoon–morning differences and late morning–mid-morning differences helps account for day-to-day variations in regional background O3 but also introduces higher relative uncertainties because four measurements (two differences) are used in the regressions. Results for all three approaches are tabulated in Table S5, by site and year. Like the regressions based on 14:00 measurements, the difference-based regressions indicate that observed slopes have increased over time (Table S5). The difference-based regressions exhibit lower slopes than the non-differenced afternoon regressions, which could be due to lesser statistical fit, or to better accounting for regional background O3, or to a combination of these factors. The difference-based regressions suggest that O3–NOz slopes increased from less than 5 : 1 in the late 1990s and early 2000s to values between 5 : 1 and 10 : 1 after 2010 (Table S5). These lower slope values are consistent with our previous results in which observed O3–NOz relationships were determined while also accounting for day-to-day variations in meteorology, which indicated that JST, YRK, and CTR O3 ∕ NOz slopes were 3.5, 5.0, and 7.1, respectively, within the range of 1 to 5 ppbv NOz, for measurements made during March–October of 2002–2011 (Blanchard et al., 2014).
A second potential effect on the temporal changes in the regression slopes could be due to changes in NO2 measurement methods (previously described); this possibility was checked by using regressions of O3 vs. HNO3 (Fig. S17). The results indicate that the relationship in Fig. 10 is not an artifact of changes in NO2 measurement methods. The record is more complete for the regressions of O3 vs. HNO3, because the HNO3 measurements were made over a longer time than the NO2 measurements (and the latter are needed for computing NOz). As shown for YRK, the year-specific slopes of 14:00 O3 vs. NOz and for O3 vs. HNO3 each increased substantially after about 2008 (Figs. 10, S17). The O3 vs. NOz and O3 vs. HNO3 regression slopes tend to level out after 2011, and possibly decrease somewhat, but variability is too high to project beyond the observed data ranges (Figs. 10, S17). Similar results are obtained for spring for JST and YRK (Figs. S18 and S19).
Our increases in year-specific slopes of O3 versus NOz potentially could be due to increasing losses of NOz species, especially HNO3, over the long-term SEARCH record. As previously noted, however, the CASTNET data show declining rates of both wet and dry nitrate deposition since the late 1990s, with no change in the ratio of deposition to emissions (Fig. 1). Therefore, the long-term slope increases cannot be attributed to increasing deposition losses of HNO3 (whether absolute or fractional). Qualitatively, the CASTNET data suggest that the observed slopes would likely be at least a factor of 2 smaller if adjusted for deposition losses. This adjustment would be comparable to the 1990s studies discussed in Sect. 4.4.2. In Fig. 9, the intercepts of year-specific regressions for 2013 approach 20 ppbv O3, which could be interpreted as a regional background O3 level relatively unaffected by local chemistry. These values are lower than those in Fig. 2 and lower than the estimated range of 48 to 59 ppbv for air transported into the Houston area. They are also lower than modeled western non-US-anthropogenic regional background O3 levels of ∼ 40–50 ppbv (Lefohn et al., 2014; Dolwick et al., 2015) but are consistent with model estimates of non-US-anthropogenic background O3 less than ∼ 30 ppbv in Atlanta (Lefohn et al., 2014). Since regression intercepts restricted to days with weather that favors O3 formation do not differ much from the intercepts of the unrestricted regressions (Fig. 9), our low intercepts for recent years do not appear to be linked to meteorological conditions that specifically favor O3 loss over formation. However, when considered over the full set of years, the O3–NOz relationships on the highest O3 days differ from those on larger subsets of the data (Fig. S14). Possibly, the intercept terms cannot be fully interpreted without additional consideration of O3 carryover in multi-day episodes, as previously noted. The intercept terms for earlier years are higher than for later years; for example, the intercepts for the YRK regressions range from 27 ± 3 to 42 ± 4 ppbv prior to 2009 (for all but 2 of these years, intercepts are 36–38 ppbv). The intercept terms for earlier years are consistent with 1997–2006 eastern US summer baseline O3 levels (32 ± 12 ppbv in the absence of continental influences) reported by Chan and Vet (2010).
Higher intercepts during early years could be due to fitting a linear regression to the upper portion or the midrange of the nonlinear relationship between O3 and NOz, as shown in Figs. 8 and S14. The nonlinearity and the downward trends in mean NOz and HNO3 mixing ratios mean that slopes of regressions computed at higher mean NOz and HNO3 mixing ratios should not be extrapolated beyond their range of applicability to the y intercept. Alternatively, the trend toward lower intercepts could reflect declining mixing ratios upwind of the study sites, consistent with documented long-term reductions of ambient O3 mixing ratios throughout the US (e.g., Chan and Vet, 2010; Lefohn et al., 2010; Paoletti et al., 2014; Simon et al., 2015; Hidy and Blanchard, 2015). As previously discussed, however, regional background O3 in the southeastern US does not appear to be trending either upward or downward.
4.4.2 Comparisons with observational and modeling studies
The preceding section demonstrates that the slopes of the regressions of O3 versus NOz increased over time and examines the potential influence of measurement artifacts, weather, deposition, and pollutant transport on the results; none of these plausible influences adequately explains why the slopes of the regressions of O3 versus NOz increased. The increasing slopes appear to indicate that relationships between O3 and NOz changed over time, yet the physical processes associated with the changes remain ambiguous. Modeling studies offer insights. Modeling process analysis by Reynolds et al. (2004) for the eastern US predicted that the number of NO cycles (i.e., the ratio of new plus recreated NO to new NO) would increase from 8 to 14 (∼ 75 %) in central (metropolitan) Atlanta and from 9 to 11 (∼ 20 %) northwest of Atlanta in response to a 60 % reduction of NOx emissions from a 1996 emissions base case. Both the modeled emission reduction (60 %; compare to Fig. 2) and the modeling subregions (central Atlanta, JST; northwest Atlanta, YRK) are directly comparable to our study period and domain. NO cycling is relevant to our regressions of O3 versus NOz because an O3 molecule is produced, with some loss, each time NO cycles through a set of reactions until NO cycling terminates in reaction products that are components of NOz (e.g., HNO3, PAN). Thus, the observed increases in the slopes of the regressions of O3 versus NOz are directionally consistent with modeling predictions but larger than the predicted 20–75 % increases in NO cycling.
The data were selected to represent periods that have consistent weather from day to day to minimize the influence of meteorological variability, and regressions of subsets of the data yield slopes and intercepts comparable to those based on all days of June and July (Fig. 9). However, the observed O3 decreases that have occurred in the region (Figs. 2 and 3) could not have occurred if O3 formation rates increased by factors of ∼ 3 to 4 (as suggested by Fig. 10) or even by a factor of 2 (Table S5), since NOx emissions declined by ∼ 60 %, or ∼ 5 % per year over 20 years (Fig. 1; Hidy et al., 2014). This consideration suggests that increased NO cycling, while likely linked with our observational results, cannot be the only factor involved. The regression slopes are nonetheless consistent with related studies when a basis for comparison exists.
The SEARCH-observed afternoon slope values of ∼ 5 : 1 prior to 2003–2007 are comparable to, or lower than, similar regression results obtained in studies during the 1990s – which showed observed summer slope values of 11 : 1 in rural Georgia in 1991 (Kleinman et al., 1994); 8.5 : 1 at rural eastern sites (Trainer et al., 1993); 7 : 1 near Birmingham, AL, in 1992 (Trainer et al., 1995); 5.7 : 1 near Nashville, TN, in 1995 (Sillman et al., 1998); and 4.7 : 1 near Nashville, TN, in 1999 (Zaveri et al., 2003) – and to modeling results and observations with composite regression slope values of 6.7 and 7.6, respectively, within the afternoon planetary boundary layer in the eastern US during the summer of 2002 (Godowitch et al., 2011). The SEARCH regression slope values prior to 2003–2007 are, as expected, higher than other 1990s values that were corrected for deposition losses, which, for example, yielded adjusted estimated values between 3 : 1 and 5 : 1 near Nashville in 1995 (Nunnermacker et al., 1998; St. John et al., 1998; Sillman et al., 1998). Our higher observed slope values after 2010 are consistent with aircraft measurements made in the southeast in August and September 2013, which show an Ox (= O3 + NO2) versus NOz slope of 17.4, and they are also consistent with model calculations, which show slopes of 14.1 to 16.7 (Travis et al., 2016). Consistent with our regressions, Travis et al. (2016) did not adjust for variations in background O3 and NOz. For comparability, we note that our O3 versus NOz regression slopes were 13.1 to 18.8 (±1.2 to 1.4) in June and July 2013 at three of four sites (25.7 ± 2.8 at the fourth site, which is the most rural in character) and our Ox versus NOz slopes were 12.0 to 18.9 (±1.2 to 1.4) at three of the four sites (25.8 ± 2.8 at the fourth site). The increase in recently observed slope values that we report is therefore supported by the 2013 data of Travis et al. (2016). Our apparently high regression slope values are comparable to observations that averaged 12.9 in ship plumes and 33.5 in assumed background marine air, as reported by Kim et al. (2016) using data from a 2002 study of ship emission plumes off the coast of southern California, though the specific conditions associated with these two studies are different from ours and thus limit the applicability of the comparisons.
The increase in regression slopes with decreasing ambient NOx and NOz is also directionally consistent with computations by Liu et al. (1987), which showed increasing OPE as NOx declines. The numerical results of the modeling calculations by Liu et al. (1987) are specific to the modeled conditions, which represented complete oxidation of VOCs over a period of months. However, increases in model-predicted NOx OPE with declining NOx result from multiple factors, such as radical reactions involving VOCs and NOx, that are pertinent to other situations (Lin et al., 1988).
In contrast to southern California, where Pollack et al. (2013) reported a shift from PAN to HNO3 production, the SEARCH data do not definitively show a changing fraction of HNO3 relative to NOy. Increasing formation of PAN (which regenerates NO2) and decreasing formation of HNO3 (which terminates cycling between NO and NO2) could facilitate O3 accumulation as ambient NOx and NOz mixing ratios continue to decline. Since the long-term SEARCH data record does not include measurements of PAN, this possible effect could not be investigated.
Observed relationships between O3 and NOy species provide insight into the potential for future O3 changes in the southeastern US. Sustained urban and rural measurements of O3 and precursors, as carried out in the SEARCH study, show O3 changes in response to emission precursor reductions in a region having unusually high emissions of natural volatile organic compounds. Anthropogenic NOx and VOC emissions are each expected to continue to decline. O3 in the southeast is sensitive to NOx, and this sensitivity is expected to strengthen with reduced anthropogenic NOx and VOC emissions. The SEARCH observations could be used for retrospective testing of model projections to enhance the credibility of the models.
This study illustrates the value of consistent, long-term measurements of O3 and reactive nitrogen made at both urban and rural sites for characterizing the response of O3 to changes in precursor emissions. In the southeastern United States, O3 production is sensitive to nitrogen oxides; long-term observations document increasing sensitivity with declining NOx emissions and ambient mixing ratios. Over a period of 14 years, the rate of change in afternoon O3 mixing ratios with respect to observed mixing ratios of NOz increased at urban and rural monitoring sites as ambient NOy mixing ratios declined. In addition, changes in the relative importance of chemical reactions that yield HNO3 compared with PAN could have played a role in altering O3 accumulation. The highest peak daily 8 h average O3 mixing ratios tended to occur in summer, but the study provides some evidence for a recent shift in the frequency of maxima to spring in some locations. Maintaining measurements of O3, NO, NO2, and NOy and adding routine observations of HNO3 and organonitrates would assist in clarifying future changes of ΔO3 ∕ ΔNOz. Long-term documentation and analysis of trends in O3 mixing ratios in relation to NOx emission reductions and decreases in ambient reactive nitrogen concentrations yields opportunities for obtaining insights about ambient O3 reductions that complement and corroborate air quality modeling predictions and add substantially to the “weight-of-evidence” approach for air quality management adopted by the US government after 2000.
The SEARCH data are available at https://www.dropbox.com/sh/o9hxoa4wlo97zpe/AACbm6LetQowrpUgX4vUxnoDa?dl=0 (ARA, 2017). EPA data are available at http://aqsdr1.epa.gov/aqsweb/aqstmp/airdata/download_files.html (EPA, 2018a) and at https://www.epa.gov/castnet (EPA, 2018b).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-8183-2018-supplement.
CLB and GMH designed the study and wrote the manuscript. CLB carried out the statistical analyses.
The authors declare that they have no conflict of interest.
The authors thank Atmospheric Research and Analysis, Inc. for collecting,
validating, and providing SEARCH data as well as John Jansen for managing the
SEARCH study. Funding for the SEARCH network was provided by Southern Company
with contributions from the Electric Power Research Institute. Southern
Company provided partial financial support for analysis of SEARCH data. We
are indebted to these sponsors for supporting this unique long-term
measurement program.
Edited by: Min
Shao
Reviewed by: David Parrish and two anonymous referees
ARA (Atmospheric Research and Analysis): available at: https://www.dropbox.com/sh/o9hxoa4wlo97zpe/AACbm6LetQowrpUgX4vUxnoDa?dl=0 (last access: 9 June 2018), 2017.
Arif, N. L. and Abdullah, A. M.: Ozone pollution and historical trends of surface background ozone level: a review, World Applied Sciences Journal, 14, 31–38, 2011.
Atkinson, R.: Atmospheric chemistry of VOCs and NOx, Atmos. Environ., 34, 2063–2101, 2000.
Berkowitz, C. M., Jobson, T., Jiang, G., Spicer, C. W., and Doskey, P. V.: Chemical and meteorological characteristics associated with rapid increases of O3 in Houston, Texas, J. Geophys. Res., 109, D10307, https://doi.org/10.1029/2003JD004141, 2004.
Berlin, S. R., Langford, A. O., Estes, M., Dong, M., and Parrish, D. D.: Magnitude, decadal changes and impact of regional background ozone transported into the greater Houston, Texas area, Environ. Sci. Technol., 47, 13985–13992, https://doi.org/10.1021/es4037644, 2013.
Blanchard, C. L., Tanenbaum, S., Hidy, G., Rasmussen, R., and Watkins, R.: NMOC, ozone and organic aerosol in the southeastern states, 1999–2007. 1. Spatial and temporal variations of NMOC mixing ratios and composition in Atlanta, Georgia, Atmos. Environ., 44, 4827–4839, 2010a.
Blanchard, C. L., Hidy, G. M., and Tanenbaum, S.: NMOC, ozone, and organic aerosol in the southeastern states, 1999–2007: 2. Ozone trends and sensitivity to NMOC emissions in Atlanta, Georgia, Atmos. Environ., 44, 4840–4849, https://doi.org/10.1016/j.atmosenv.2010.07.030, 2010b.
Blanchard, C. L., Hidy, G. M., Tanenbaum, S., Edgerton, E. S., and Hartsell, B. E.: The Southeastern Aerosol Research and Characterization (SEARCH) study: Spatial variations and chemical climatology, 1999–2010, J. Air Waste Manage., 63, 260–275, https://doi.org/10.1080/10962247.2012.749816, 2013.
Blanchard, C. L., Tanenbaum, S., and Hidy, G.: Ozone in the southeastern United States: an observation-based model using measurements from the SEARCH network, Atmos. Environ., 48, 192–200, 2014.
Chameides, W. and Cowling, E.: The State of the Southern Oxidants Study: Policy Relevant Findings in Ozone Pollution Research, 1988–1994, Southern Oxidant Study, College of Forest Resources, North Carolina State University, Raleigh, NC, 1995.
Chameides, W., Lindsay, R., Richardson, J., and Kiang, C.: The role of biogenic hydrocarbons in urban photochemical smog: Atlanta as a case study, Science, 24, 1473–1475, 1988.
Chan, E.: Regional ground-level ozone trends in the context of meteorological influences across Canada and the eastern United States from 1997 to 2006, J. Geophys. Res.-Atmos., 114, D05301, https://doi.org/10.1029/2008JD010090, 2009.
Chan, E. and Vet, R. J.: Baseline levels and trends of ground level ozone in Canada and the United States, Atmos. Chem. Phys., 10, 8629–8647, https://doi.org/10.5194/acp-10-8629-2010, 2010.
Cooper, O., Gao, R.-S., Tarasick, D., Leblanc, T., and Sweeney, C.: Long-term ozone trends at rural ozone monitoring sites across the United States, 1990–2010, J. Geophys. Res.-Atmos., 117, D22307, https://doi.org/10.1029/2012JD018261, 2012.
Cooper, O., Parrish, D., Ziemke, J., Balashov, N., Cupeiro, M., Galbally, I., Gilge, S., Horowitz, I., Jensen, N., Larmarque, J., Naik, N., Oltmans, S., Schwab, J., Shindell, D., Thompson, A., Thouret, V., Wang, Y., and Zhinden, R.: Global distribution and trends of tropospheric ozone: an observation-based review, Elementa, 2, 1–28, https://doi.org/10.12952/journal.elementa.000029, 2014.
Cristofanelli, P. and Bonasoni, P.: Background ozone in the southern Europe and Mediterranean area: influence of the transport processes, Environ. Pollut., 157, 1399–1406, https://doi.org/10.1016/j.envpol.2008.09.017, 2009.
de Gouw, J. A., Parrish, D. D., Frost,G. J., and Trainer, M.: Reduced emissions of CO2, NOx, and SO2 from U.S. power plants owing to switch from coal to natural gas with combined cycle technology, Earths Future, 2, 75–82, https://doi.org/10.1002/2013EF000196, 2014.
Dolwick, P., Akhtar, F., Baker, K. R., Possiel, N., and Simon, H.: Comparison of background ozone estimates over the western United States based on two separate model methodologies, Atmos. Environ, 109, 282–296, https://doi.org/10.1016/j.atmosenv.2015.01.005, 2015.
Edgerton, E. S., Saylor, R. D., Hartsell, B. E., Jansen, J. J., and Hansen, D. A.: Ammonia and ammonium measurements from the southeastern United States, Atmos. Environ., 41, 3339–3351, 2007.
EPA: Pre-Generated Data Files, available at: https://aqs.epa.gov/aqsweb/airdata/download_files.html, last access: 9 June 2018a.
EPA: Clean Air Status and Trends Network (CASTNET), available at: https://www.epa.gov/castnet, last access: 9 June 2018b.
Fiore, A., Oberman, J. T., Lin, M. Y., Zhang, L., Clifton, O. E., Jacob, D. J., Naik, V., Horowitz, L. W., Pinto, J. P., and Milly, G.: Estimating North American background ozone in U.S. surface air with two independent global models: variability, uncertainties, and recommendations, Atmos. Environ., 96, 284–300, 2014.
Frost, G. J., Trainer, M., Allwine, G., Buhr, M. P., Calvert, J. G., Cantrell, C. A., Fehsenfeld, F. C., Goldan, P. D., Herwehe, J., Hubler, G., Kuster, W. C., Martin, R., McMillen, R. T., Montzka, S. A., Norton, R. B., Parrish, D. D., Ridley, B. A., Shetter, R. E., Walega, J. G., Watkins, B. A., Westberg, H. H., and Williams, E. J.: Photochemical ozone production in the rural southeastern United States during the 1990 Rural Oxidants in the Southern Environment (ROSE) program, J. Geophys. Res.-Atmos., 103, 22491–22508, 1998.
Fujita, E., Stockwell, W., Campbell, D., Keisslar, R., and Lawson, D.: Evolution of the magnitude and spatial extent of the weekend ozone effect in California's South Coast Air Basin, 1981–2000, J. Air Waste Manage., 53, 802–815, 2003.
Fujita, E., Campbell, D. E., Stockwell, W., Saunders, E., Fitzgerald, R., and Perea, R.: Projected ozone trends and changes in the ozone-precursor relationship in the South Coast Air Basin in response to varying reductions of precursor emissions, J. Air Waste Manage., 66, 201–214, 2015.
Godowitch, J. M., Gilliam, R. C., and Rao, S. T.: Diagnostic evaluation of ozone production and horizontal transport in a regional photochemical air quality modeling system, Atmos. Environ., 45, 3977–3987, 2011.
Griffin, R. J., Johnson, C. A., Talbot, R. W., Mao, H., Russo, R. S., Zhou, Y., and Sive, B. C.: Quantification of ozone formation metrics at Thompson Farm during the New England Air Quality Study (NEAQS) 2002, J. Geophys. Res.-Atmos., 109, D24302, https://doi.org/10.1029/2004JD005344, 2004.
Hansen, D. A., Edgerton, E. S., Hartsell, B. E., Jansen, J. J., Hidy, G. M., Kandasamy, K., and Blanchard, C. L.: The Southeastern Aerosol Research and Characterization Study (SEARCH): 1. Overview, J. Air Waste Manage., 53, 1460–1471, 2003.
Hansen, D. A., Edgerton, E., Hartsell, B., Jansen, J., Burge, H., Koutrakis, P., Rogers, C., Suh, H., Chow, J., Zielinska, B., McMurry, P., Mulholland, J., Russell, A., and Rasmussen, R.: Air quality measurements for the aerosol research and inhalation epidemiology study, J. Air Waste Manage., 56, 1445–1458, 2006.
He, H., Hembeck, L., Hosley, K. M., Canty, T. P., Salawitch, R. J., and Dickerson, R. R.: High ozone concentrations on hot days: The role of electric power demand and NOx emissions, Geophys. Res. Lett., 40, 5291–5294, https://doi.org/10.1002/grl.50967, 2013.
Hidy, G. M. and Blanchard, C. L.: Precursor reductions and ground-level ozone in the continental U.S., J. Air Waste Manage., 65, 1261–1282, https://doi.org/10.1080/10962247.2015.1079564, 2015.
Hidy, G. M., Blanchard, C. L., Baumann, K., Edgerton, E., Tanenbaum, S., Shaw, S., Knipping, E., Tombach, I., Jansen, J., and Walters, J.: Chemical climatology of the southeastern United States, 1999–2013, Atmos. Chem. Phys., 14, 11893–11914, https://doi.org/10.5194/acp-14-11893-2014, 2014.
Hirsch, A. I., Munger, J. W., Jacob, D. J., Horowitz, L. W., and Goldstein, A. H.: Seasonal variation of the ozone production efficiency per unit NOx at Harvard Forest, Massachusetts, J. Geophys. Res.-Atmos., 101, 12659–12666, https://doi.org/10.1029/96JD00557, 1996.
Hudman, R. C., Jacob, D. J., Turquety, S., Leibensperger, E. M., Murray, L. T., Wu, S., Gilliland, A. B., Avery, M., Bertram, T. H., Brune, W., Cohen, R. C., Dibb, Flocke, F. M., Fried, A., Holloway, J., Neuman, J. A., Orville, R., Perring, A., Ren, X., Sachse, G. W., Singh, H. B., Swanson, A., and Wooldridge, P. J.: Surface and lightning sources of nitrogen oxides over the United States: Magnitudes, chemical evolution, and outflow, J. Geophys. Res.-Atmos., 112, D12S05, https://doi.org/10.1029/2006JD007912, 2007.
Jacob, D. J., Horowitz, L. W., Munger, J. W., Heikes, B. G., Dickerson, R. R., Artz, R. S., and Keene, W. C.: Seasonal transition from NOx- to hydrocarbon-limited conditions for ozone production over the Eastern United States in September, J. Geophys. Res.-Atmos., 100, 9315–9324, 1995.
Johnson, M., Kuang, S., Wang, L., and Newchurch, M.: Evaluating summer-time ozone enhancement events in the southeast United States, Atmosphere, 7, 108, https://doi.org/10.3390/atmos7080108, 2016.
Kasibhatla, P., Chameides, W. L., Saylor, R. D., and Olerud, D.: Relationships between regional ozone pollution and emissions of nitrogen oxides in the eastern United States, J. Geophys. Res.-Atmos., 103, 22663–22669, https://doi.org/10.1029/98JD01639, 1998.
Kim, H. S., Kim, Y. H., Han, K. M., Kim,J., and Song, C. H.: Ozone production efficiency of a ship-plume: ITCT 2K2 case study, Chemosphere, 143, 17–23, 2016.
Kleinman, L., Lee, Y.-N., Springston, S. R., Nunnermacker, L., Zhou, X., Brown, R., Hallock, K., Klotz, P., Leahy, D., Lee, J. H., and Newman, L.: Ozone formation at a rural site in the southeastern United States, J. Geophys. Res.-Atmos., 99, 3469–3482, https://doi.org/10.1029/93JD02991, 1994.
Kleinman, L. I., Daum, P. H., Lee, Y. N., Senum, G. I., Springston, S. R., Wany, J., Berkowitz, C., Hubbe, J., Zaveri, R. A., Brechtel, F. J., Jayne, J., Onasch, T. B., and Worsnop, D.: Aircraft observations of aerosol composition and ageing in New England and mid-Atlantic states during the summer 2002 New England Air Quality Study field campaign, J. Geophys. Res.-Atmos., 112, D09310, https://doi.org/10.1029/2006JD007786, 2007.
Langford, A. O., Senff, C. J., Alvarez II, R. J., Brioude, J., Cooper, O. R., Holloway, J. S., Lin, M. Y., Marchbanks, R. D., Pierce, R. B., Sandberg, S. P., Weickmann, A. M., and Williams, E. J.: An overview of the 2013 Las Vegas Ozone Study (LVOS): Impact of stratospheric intrusions and long-range transport on surface air quality, Atmos. Environ., 109, 305–322, 2015.
Lefohn, A., Shadwick, D., and Oltmans, S.: Characterizing changes in surface ozone levels in metropolitan and rural areas in the United States for 1980–2008 and 1994–2008, Atmos. Environ., 44, 5199–5210, 2010.
Lefohn, A., Emery, C., Shadwick, D., Wernli, H., Jung, J., and Oltmans, S.: Estimates of background surface ozone mixing ratios in the United States based on model-derived source apportionment, Atmos. Environ., 84, 275–288, https://doi.org/10.1016/j.atmosenv.2013.11.033, 2014.
Lin, M., Fiore, A. M., Cooper, O. R., Horowitz, L. W., Langford, A. O., Levy, H., Johnson, B. J., Naik, V., Oltmans, S. J., and Senff, C. J.: Springtime high surface ozone events over the western United States: Quantifying the role of stratospheric intrusions, J. Geophys. Res.-Atmos., 117, D00V22, https://doi.org/10.1029/2012JD018151, 2012.
Lin, M., Horowitz, L. W., Payton, R., Fiore, A. M., and Tonnesen, G.: US surface ozone trends and extremes from 1980 to 2014: quantifying the roles of rising Asian emissions, domestic controls, wildfires, and climate, Atmos. Chem. Phys., 17, 2943–2970, https://doi.org/10.5194/acp-17-2943-2017, 2017.
Lin, X., Trainer, M., and Liu, S. C.: On the nonlinearity of the tropospheric ozone production, J. Geophys. Res.-Atmos., 93, 15879–15888, 1988.
Liu, S. C., Trainer, M., Fehsenfeld, F. C., Parrish, D. D., Williams, E. J., Fahey, D. W., Hubler, G., and Murphy, P. C.: Ozone production in the rural troposphere and the implications for regional and global ozone distributions, J. Geophys. Res., 92, 4191–4207, 1987.
Logan, J.: Tropospheric ozone: seasonal behavior, trends, and anthropogenic influence., J. Geophys. Res., 90, 10463–10482, 1985.
McDuffie, E. E., Edwards, P. M., Gilman, J. B., Lerner, B. M., Dubé, W. P., Trainer, M., Wolfe, D. E., Angevine, W. M., deGouw, J., Williams, E. J., Tevlin, A. G., Murphy, J. G., Fischer, E. V., McKeen, S., Ryerson, T. B., Peischl, J., Holloway, J. S., Aikin, K., Langford, A. O., Senff, C. J., Alvarez II, R. J., Hall, S. R., Lantz, K. O., and Brown, S. S.: Influence of oil and gas emissions on summertime ozone in the Colorado Northern Front Range, J. Geophys. Res.-Atmos., 121, 8712–8729, https://doi.org/10.1002/2016JD025265, 2016.
Meagher, J., Cowling, E., Fehsenfeld, F., and Parkhurst, W.: Ozone formation and transport in southeasterm United States: overview of the SOS Nashville/Middle Tennessee Study, J. Geophys. Res., 103, 22213–22223, 1998.
Naja, M., Akimoto, H., and Staehelin, J.: Ozone in background and photochemically aged air over central Europe: Analysis of long-term ozonesonde data from Hohenpeissenberg and Payerne, J. Geophys. Res., 108, 4063, https://doi.org/10.1029/2002JD002477, 2003.
NARSTO: An Assessment of Tropospheric Ozone Pollution, Report 1000040, NARSTO, Pasco, WA (also available from EPRI, Palo Alto, CA), 2000.
Neuman, J. A., Nowak, J. B., Zheng, W., Flocke, F., Ryerson, T. B., Trainer, M., Holloway, J. S., Parrish, D. D., Frost, G. J., Peischl, J., Atlas, E. L., Bahreini, R., Wollny, A. G., and Fehsenfeld, F. C.: Relationship between photochemical ozone production and NOx oxidation in Houston, Texas, J. Geophys. Res.-Atmos., 114, D00F08, https://doi.org/10.1029/2008JD011688, 2009.
Neuman, J. A., Trainer, M., Brown, S. S., Min, K.-E., Nowak, J. B., Parrish, D. D., Peischl, J., Pollack, I. B., Roberts, J. M., Ryerson, T. B., and Veres, P. R.: HONO emission and production determined from airborne measurements over the Southeast U.S., J. Geophys. Res.-Atmos., 121, 9237–9250, https://doi.org/10.1002/2016JD025197, 2016.
Newchurch, M., Ayoub, M., Oltmans, S., Johnson, B., and Schmidlin, F.: Vertical distributions of ozone at four sites in the United States, J. Geophys. Res., 208, 4031, https://doi.org/10.1029/2002JD002059, 2003.
NOAA (National Oceanic and Atmospheric Administration): ESRL/GMD FTP Data Finder, available at: ftp://aftp.cmdl.noaa.gov/data/ozwv/Ozonesonde/ (last access: 9 June 2018), 2017.
NRC (National Research Council): Rethinking the Ozone Problem in Urban and Regional Air Pollution, National Academy Press, Washington, D.C., 489 pp., 1991.
Nunnermacker, L. J., Imre, D., Daum, P. H., Kleinman, L., Lee, Y.-N., Lee, J. H., Springston, S. R., Newman, L., Weinstein-Lloyd, J., Luke, W. T., Banta, R., Alvarez, R., Senff, C., Sillman, S., Holdren, M., Keigley, G. W., and Zhou, X.: Characterization of the Nashville urban plume on July 3 and July 18, 1995, J. Geophys. Res.-Atmos., 103, 28129–28148, https://doi.org/10.1029/98JD01961, 1998.
Oltmans, S., Lefohn, A., Harris, J., and Shadwick, D.: Background ozone levels of air entering the west coast of the US and assessment of longer-term changes, Atmos. Environ., 42, 6020–6038, 2008.
Oltmans, S., Lefohn, A., Shadwick, D., Harris, J., Scheel, H., Galbally, I., Tarasick, D., Johnson, B., Brunke, E., Claude, H., Zeng, G., Nichol, S., Schmidlin, F., Davies, J., Cuevas, E., Redondas, A., Naoe, H., Kakano, T., and Kawasato, T.: Recent tropospheric ozone changes – a pattern dominated by slow or no growth, Atmos. Environ., 67, 331–351, 2013.
Ordóñez, C., Brunner, D., Staehelin, J., Hadjinicolaou, P., Pyle, J. A., Jonas, M., Wernli, H., and Prévôt, A. S. H.: Strong influence of lowermost stratospheric ozone on lower tropospheric background ozone changes over Europe, Geophys. Res. Lett., 34, L07805, https://doi.org/10.1029/2006GL029113, 2007.
Paoletti, E., De Marco, A., Beddows, D. C. S., Harrison, R. M., and Manning, W. J.: Ozone levels in European and USA cities are increasing more than at rural sites, while peak values are decreasing, Environ. Pollut., 192, 295–299, 2014.
Parrish, D. D., Young, L. M., Newman, M. H., Aikin, K. C., and Ryerson, T. B.: Ozone design values in Southern California's air basins: Temporal evolution and U.S. background contribution, J. Geophys. Res., 122, 11166–11182, https://doi.org/10.1002/2016JD026329, 2017a.
Parrish, D. D., Petropavlovskikh, I., and Oltmans, S. J.: Reversal of long-term trend in baseline ozone concentrations at the North American West Coast, Geophys. Res. Lett., 44, 10675–10681, https://doi.org/10.1002/2017GL074960, 2017b.
Pollack, I., Ryerson, T., Trainer, M., Neuman, J., Roberts, J., and Parrish, D.: Trends in ozone its precursors and related secondary oxidation products in Los Angeles, California: a synthesis of measurements from 1960–2010, J. Geophys. Res.-Atmos., 118, 5891–5911, 2013.
Reynolds, S., Blanchard, C. L., and Ziman, S.: Understanding the effectiveness of precursor reductions in lowering 8-hour ozone mixing ratios, J. Air Waste Manage., 53, 195–205, 2003.
Reynolds, S., Blanchard, C. L., and Ziman, S.: Understanding the effectiveness of precursor reductions in lowering the 8-hr. O3 concentration: part II-eastern United States, J. Air Waste Manage., 54, 1452–1470, 2004.
Ryerson, T. B., Andrews, A. E., Angevine, W. M., Bates, T. S., Brock, C. A., Cairns, B., Cohen, R. C., Cooper, O. R., de Gouw, J. A., Fehsenfeld, F. C., Ferrare, R. A., Fischer, M. L., Flagan, R. C., Goldstein, A. H., Hair, J. W., Hardesty, R. M., Hostetler, C. A., Jimenez, J. L., Langford, A. O., McCauley, E., McKeen, S. A., Molina, L. T., Nenes, A., Oltmans, S. J., Parrish, D. D., Pederson, J. R., Pierce, R. B., Prather, K., Quinn, P. K., Seinfeld, J. H., Senff, C. J., Sorooshian, A., Stutz, J., Surratt, J. D., Trainer, M., Volkamer, R., Williams, E. J., and Wofsy, S. C.: The 2010 California Research at the Nexus of Air Quality and Climate Change (CalNex) field study, J. Geophys. Res.-Atmos., 118, 5830–5866, https://doi.org/10.1002/jgrd.50331, 2013.
Saylor, R. D., Edgerton, E. S., Hartsell, B. E., Baumann, K., and Hansen, D. A.: Continuous gaseous and total ammonia measurements from the southeastern aerosol research and characterization (SEARCH) study, Atmos. Environ., 44, 4994–5004, 2010.
Schere, K. and Hidy, G. M.: Foreword: NARSTO critical reviews, Atmos. Environ., 34, 1853–1860, 2000.
Schnell, R. C., Oltmans, S. J., Neely, R. R., Endres, M. S., Molenar, J. V., and White, A. B.: Rapid photochemical production of ozone at high concentrations in a rural site during winter, Nat. Geosci., 2, 120–122, https://doi.org/10.1038/ngeo415, 2009.
Seigneur, C. and Dennis, R.: Atmospheric modeling, in: Technical Challenges of Multipollutant Air Quality Management, edited by: Hidy, G. M., Brook, J. R., Demerjian, K. L., Molina, L. T., Pennell, W. T., and Scheffe, R. D., Springer, New York, 2011.
Seinfeld, J. H.: Atmospheric Chemistry and Physics of Air Pollution, John Wiley and Sons, New York, 1986.
Sillman, S., He, D., Pippin, M., Daum, P. H., Imre, D. G., Kleinman, L. I., and Lee, J. H.: Model correlations for ozone, reactive nitrogen, and peroxides for Nashville in comparison with measurements: Implications for O3-NOx-hydrocarbon chemistry, J. Geophys. Res.-Atmos., 103, 22629–22644, 1998.
Simon, H., Reff, A., Wells, B., Xing, J., and Frank, N.: Ozone trends across the United States over a period of decreasing NOx and VOC emissions, Environ. Sci. Technol., 49, 186–195, https://doi.org/10.1021/es504514z, 2015.
Singh, H.: Reactive nitrogen in the troposphere: chemistry and transport of NOx and PAN, Environ. Sci. Technol., 21, 320–327, 1987.
Singh, H. B. and Hanst, P. L.: Peroxyacetyl nitrate (PAN) in the unpolluted atmosphere: an important reservoir for nitrogen oxides, Geophys. Res. Lett., 8, 941–944, 1981.
Solberg, S., Derwent, R. G., Hov, Ø., Langner, J., and Lindskog, A.: European abatement of surface ozone in a global perspective, Ambio, 34, 47–53, https://doi.org/10.1579/0044-7447-34.1.47, 2005.
Solomon, P., Cowling, E., Hidy, G., and Furness, C.: Comparison of scientific findings from major ozone field studies in North America and Europe, Atmos. Environ., 34, 1885–1920, 2000.
St. John, J. C., Chameides, W. L., and Saylor, R.: Role of anthropogenic NOx and VOC as ozone precursors: A case study from the SOS Nashville/Middle Tennessee Ozone Study, J. Geophys. Res.-Atmos., 103, 22415–22423, 1998.
Starn, T. K., Shepson, P. B., Bertman, S. B., White, J. S., Splawn, B. G., Riemer, D. D., Zika, R. G., and Olszyna, K.: Observations of isoprene chemistry and its role in ozone production at a semirural site during the 1995 Southern Oxidants Study, J. Geophys. Res., 103, 22425–22435, https://doi.org/10.1029/98JD01279, 1998.
Trainer, M., Parrish, D. D., Buhr, M. P., Norton, R., Fehsenfeld, F., Anlauf, K., Bottenheim, J., Tang, Y., Weibe, H., Roberts, J., Tanner, R., Newman, L., Bowersox, V., Meagher, J., Olszyna, K., Rodgers, M., Wang, T., Berresheim, H., Demerjian, K., and Roychowdhury, U.: Correlation of ozone with NOy in photochemically aged air, J. Geophys. Res.-Atmos., 98, 2917–2925, 1993.
Trainer, M., Ridley, B. A., Buhr, M. P., Kok, G., Walega, J., Hübler, G., Parrish, D. D., and Fehsenfeld, F. C.: Regional ozone and urban plumes in the southeastern United States: Birmingham, A case study, J. Geophys. Res.-Atmos., 100, 18823–18834, https://doi.org/10.1029/95JD01641, 1995.
Trainer, M., Parrish, D., Goldan, P., Roberts, P. J., and Fehsenfeld, F.: Review of observation-based analysis of regional factors influencing ozone concentration, Atmos. Environ., 34, 2045–2062, 2000.
Travis, K. R., Jacob, D. J., Fisher, J. A., Kim, P. S., Marais, E. A., Zhu, L., Yu, K., Miller, C. C., Yantosca, R. M., Sulprizio, M. P., Thompson, A. M., Wennberg, P. O., Crounse, J. D., St. Clair, J. M., Cohen, R. C., Laughner, J. L., Dibb, J. E., Hall, S. R., Ullmann, K., Wolfe, G. M., Pollack, I. B., Peischl, J., Neuman, J. A., and Zhou, X.: Why do models overestimate surface ozone in the Southeast United States?, Atmos. Chem. Phys., 16, 13561–13577, https://doi.org/10.5194/acp-16-13561-2016, 2016.
University of Alabama Huntsville (UAH): Huntsville ozonesonde station, available at: http://www.nsstc.uah.edu/atmchem/about_ozonesonde.html (last access: 2 October 2017), 2017.
U.S. EPA: Regional and Seasonal Analysis of North American Background Ozone Estimated from Two Studies, available at: https://www3.epa.gov/ttn/naaqs/standards/ozone/data/20120814BackgroundOzone.pdf (last access: 9 June 2018), 2012.
U.S. EPA: Health Risk and Exposure Assessment for Ozone Final Report, EPA-452/R-14-004a, available at: http://www.epa.gov/ttn/naaqs/standards/ozone/data/20140829healthrea.pdf, (last access: 19 August 2015), 2014.
U.S. EPA: Ozone (O3) Standards – Documents from Current Review – Risk and Exposure Assessments, available at: https://www.epa.gov/naaqs/ozone-o3-standards-risk-and-exposure-assessments-current-review (last access: 9 June 2018), 2015a.
U.S. EPA: Ozone (O3) Standards – Table of Historical Ozone NAAQS, available at: http://www.epa.gov/ttn/naaqs/standards/ozone/s_o3_history.html (last access: 19 August 2015), 2015b.
U.S. EPA: Environmental Protection Agency 40 CFR Parts 50, 51, 52, 53 and 58 [EPA-HQ-OAR-2008-0699; FRL-9918-43-OAR] RIN 2060-AP38, National Ambient Air Quality Standards for Ozone, available at: http://www.epa.gov/airquality/ozonepollution/pdfs/20141125proposal.pdf (last access: 19 August 2015), 2015c.
U.S. EPA: National Trends in Ozone Levels, available at: http://www.epa.gov/airtrends/ozone.html (last access: 19 August 2015), 2015d.
U.S. EPA: Air Quality Trends, available at: http://www.epa.gov/airtrends/aqtrends.html#comparison (last access: 19 August 2015), 2015e.
U.S. EPA: AirData: Download Data Files, available at: http://aqsdr1.epa.gov/aqsweb/aqstmp/airdata/download_files.html (last access: 15 March 2017), 2016a.
U.S. EPA: Clean Air Status and Trends Network (CASTNET), available at: https://www.epa.gov/castnet (last access: 24 March 2016), 2016b.
U.S. EPA: Air Markets Program Data, available at: http://ampd.epa.gov/ampd/ (last access: 12 September 2016), 2016c.
U.S. EPA: Air Pollutant Emission Trends Data, available at: https://www.epa.gov/air-emissions-inventories/air-pollutant-emissions-trends-data (last access: 25 September 2016), 2016d.
Warneke, C., Trainer, M., de Gouw, J. A., Parrish, D. D., Fahey, D. W., Ravishankara, A. R., Middlebrook, A. M., Brock, C. A., Roberts, J. M., Brown, S. S., Neuman, J. A., Lerner, B. M., Lack, D., Law, D., Hübler, G., Pollack, I., Sjostedt, S., Ryerson, T. B., Gilman, J. B., Liao, J., Holloway, J., Peischl, J., Nowak, J. B., Aikin, K. C., Min, K.-E., Washenfelder, R. A., Graus, M. G., Richardson, M., Markovic, M. Z., Wagner, N. L., Welti, A., Veres, P. R., Edwards, P., Schwarz, J. P., Gordon, T., Dube, W. P., McKeen, S. A., Brioude, J., Ahmadov, R., Bougiatioti, A., Lin, J. J., Nenes, A., Wolfe, G. M., Hanisco, T. F., Lee, B. H., Lopez-Hilfiker, F. D., Thornton, J. A., Keutsch, F. N., Kaiser, J., Mao, J., and Hatch, C. D.: Instrumentation and measurement strategy for the NOAA SENEX aircraft campaign as part of the Southeast Atmosphere Study 2013, Atmos. Meas. Tech., 9, 3063–3093, https://doi.org/10.5194/amt-9-3063-2016, 2016.
Wiedinmyer, C., Tie, X., Guenther, A., Neilson, R., and Granier, C.: Future changes in biogenic isoprene emissions: how might they affect regional and global atmospheric chemistry?, Earth Interact., 10, 1–19, https://doi.org/10.1175/EI174.1, 2006.
Williams, E. J., Baumann, K., Roberts, J. M., Bertman, S. B., Norton, R. B., Fehsenfeld, F. C., Springston, S. R., Nunnermacker, L. J., Newman, L., Olszyna, K., Meagher, J., Hartsell, B., Edgerton, E., Pearson, J. R., and Rodgers, M. O.: Intercomparison of ground-based NOy measurement techniques, J. Geophys. Res., 103, 22261–22280, 1998.
Wilson, R. C., Fleming, Z. L., Monks, P. S., Clain, G., Henne, S., Konovalov, I. B., Szopa, S., and Menut, L.: Have primary emission reduction measures reduced ozone across Europe? An analysis of European rural background ozone trends 1996–2005, Atmos. Chem. Phys., 12, 437–454, https://doi.org/10.5194/acp-12-437-2012, 2012.
Xing, J., Pleim, J., Mathur, R., Pouliot, G., Hogrefe, C., Gan, C.-M., and Wei, C.: Historical gaseous and primary aerosol emissions in the United States from 1990 to 2010, Atmos. Chem. Phys., 13, 7531–7549, https://doi.org/10.5194/acp-13-7531-2013, 2013.
Zaveri, R. A., Berkowitz, C. M., Kleinman, L. I., Springston, S. R., Doskey, P. V., Lonneman, W. A., and Spicer, C. W.: Ozone production efficiency and NOx depletion in an urban plume: Interpretation of field observations and implications for evaluating O3-NOx-VOC sensitivity, J. Geophys. Res.-Atmos., 108, 4436, https://doi.org/10.1029/2002JD003144, 2003.
Zhang, L., Jacob, D. J., Downey, N. V., Wood, D. A., Blewitt, D., Carouge, C. C., van Donkelaar, A., Jones, D. B. A., Murray, L. T., and Wang, Y.: Improved estimate of the policy-relevant background ozone in the United States using the GEOS-Chem global model with 1/2∘ × 2/3∘ horizontal resolution over North America, Atmos. Environ., 45, 6769–6776, https://doi.org/10.1016/j.atmosenv.2011.07.054, 2011.