the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 09 May 2018
| 09 May 2018
NO2-initiated multiphase oxidation of SO2 by O2 on CaCO3 particles
Ting Yu
Defeng Zhao
Xiaojuan Song
The reaction of SO2 with NO2 on the surface of aerosol particles has been suggested to be important in sulfate formation during severe air pollution episodes in China. However, we found that the direct oxidation of SO2 by NO2 was slow and might not be the main reason for sulfate formation in ambient air. In this study, we investigated the multiphase reaction of SO2 with an O2 ∕ NO2 mixture on single CaCO3 particles using Micro-Raman spectroscopy. The reaction converted the CaCO3 particle to a Ca(NO3)2 droplet, with CaSO4 • 2H2O solid particles embedded in it, which constituted a significant fraction of the droplet volume at the end of the reaction. The reactive uptake coefficient of SO2 for sulfate formation was on the order of 10−5, which was higher than that for the multiphase reaction of SO2 directly with NO2 by 2–3 orders of magnitude. According to our observations and the literature, we found that in the multiphase reaction of SO2 with the O2 ∕ NO2 mixture, O2 was the main oxidant of SO2 and was necessary for radical chain propagation. NO2 acted as the initiator of radical formation, but not as the main oxidant. The synergy of NO2 and O2 resulted in much faster sulfate formation than the sum of the reaction rates with NO2 and with O2 alone. We estimated that the multiphase oxidation of SO2 by O2 initiated by NO2 could be an important source of sulfate and a sink of SO2, based on the calculated lifetime of SO2 regarding the loss through the multiphase reaction versus the loss through the gas-phase reaction with OH radicals. Parameterization of the reactive uptake coefficient of the reaction observed in our laboratory for further model simulation is needed, as well as an integrated assessment based on field observations, laboratory study results, and model simulations to evaluate the importance of the reaction in ambient air during severe air pollution episodes, especially in China.
- Article
(934 KB) - Companion paper
-
Supplement
(224 KB) - BibTeX
- EndNote





It has been suggested that multiphase or heterogeneous oxidation of SO2 potentially plays an important role in sulfate formation in the atmosphere (Seinfeld and Pandis, 2006). During the severe pollution episodes that occur frequently in China, high sulfate concentrations cannot be explained by the gas-phase oxidation of SO2 and its well known aqueous chemistry (Zheng et al., 2015a; Cheng et al., 2016), highlighting the role of underappreciated heterogeneous oxidation or multiphase pathways.
Recently, the multiphase oxidation of SO2 by NO2 has been introduced in air quality model simulations to explain the discrepancy between the modeled and observed sulfate concentration during severe pollution episodes in China (Cheng et al., 2016; Gao et al., 2016; Wang et al., 2016; Xue et al., 2016), despite the uncertainties in the kinetic parameters for SO2 oxidation and in the pH value of aerosol particles in China (Wang et al., 2016; Cheng et al., 2016; Liu et al., 2017; Guo et al., 2017). However, according to our recently published results (Zhao et al., 2018), the direct oxidation of SO2 by NO2 could not contribute significantly to sulfate formation in the atmosphere because the reactive uptake coefficient of SO2 for sulfate formation due to direct oxidation by NO2 is very low (∼ 10−8).
Although the contribution of the direct oxidation of SO2 by NO2 to sulfate formation is not significant, NO2 may be involved in other oxidation pathways of SO2. It has been reported that the reaction of NO2 with SO and HSO in the bulk aqueous phase can form the SO radical, which can further react with O2 and produce a series of radicals that oxidize S(IV) species (Littlejohn et al., 1993). The reaction pathway may result in a fast SO2 oxidation due to the potential synergy of NO2 and O2.
Despite such a reaction mechanism for SO2 oxidation being proposed, its role in SO2 oxidation in the ambient atmosphere is not well established. Most previous studies have focused on the direct reaction of SO2 with NO2, including the determination of its rate constant (Lee and Schwartz, 1983; Clifton et al., 1988; Shen and Rochelle, 1998; Spindler et al., 2003; Nash, 1979; Huie and Neta, 1986). According to the reaction products and their reported yields (Lee and Schwartz, 1983; Clifton et al., 1988), the overall reaction equations of the direct reaction of SO2 with NO2 are as follows:
and the reactions are proposed to proceed via NO2–S(IV) adduct complexes (Clifton et al., 1988).
Additionally, NO2–S(IV) adduct complexes may decompose as follows (Spindler et al., 2003):
However, studies of the oxidation rate of SO2 at the O2 concentrations relevant to the ambient atmosphere and the potential influence of the synergy of NO2 and O2 on the oxidation rate are very limited (Turšič et al., 2001; He et al., 2014), except for a few studies which investigated SO2 oxidation in the presence of NO2 as well as O2 (Littlejohn et al., 1993; Shen and Rochelle, 1998; Santachiara et al., 1990). Moreover, previous studies have mainly focused on the reaction in bulk solution and only few studies have investigated the oxidation of SO2 by NO2 on aerosol particles (Santachiara et al., 1990, 1993). On aerosol particles, water activity, pH, ionic strength, the presence of other compounds or ions, and the role of the particle surface are different from dilute bulk solution and may affect the reaction process and reaction rate. Therefore, further studies of the multiphase reaction of SO2 with O2 ∕ NO2 mixtures on aerosol particles are required to determine the kinetic parameters and the mechanism of the reaction.
In this study, we investigated the multiphase reaction of SO2 with O2 in the presence of NO2 on CaCO3 particles. We quantified the reactive uptake coefficient of SO2 due to the reaction with an O2 ∕ NO2 ∕ H2O mixture. Based on our observations and the existing literature, we further discussed the reaction mechanism. Furthermore, we estimated the role of the multiphase oxidation of SO2 by O2 in the presence of NO2 in the atmosphere.
The experiments were conducted using a flow reaction system and the setup is shown in Fig. S1 in the Supplement. The experimental setup and procedure used have been described in detail in previous studies (Zhao et al., 2018, 2011; Liu et al., 2008). A gas mixture of NO2, SO2, O2, N2, and water vapor reacted with particles deposited on a substrate in the flow reaction cell. The concentrations of SO2 and NO2 were controlled using mass flow controllers by varying the flow rates of SO2 (2000 ppm in high purity N2, National Institute of Metrology P.R. China), NO2 (1000 ppm in high purity N2, Messer, Germany), and synthetic air (20 % O2 (high purity grade: 99.999 %, Beijing Haikeyuanchang Practical Gas Co., Ltd.) and 80 % N2 (high purity grade: 99.999 %, Beijing Haikeyuanchang Practical Gas Co., Ltd.)). Relative humidity (RH) was controlled by regulating the flow rates of reactant gases, dry synthetic air, and humidified synthetic air. Humidified synthetic air was prepared by bubbling synthetic air through fritted glass in water. In some experiments, the O2 concentrations were varied by regulating the mixing ratios of O2 and N2 to investigate the effect of O2. SO2 ∕ O2 ∕ NO2 ∕ H2O mixtures flowed through the reaction cell and reacted with individual stationary CaCO3 particles, which were deposited on a Teflon® FEP film substrate annealed to a silicon wafer. RH and temperature were measured using a hygrometer (HMT100, Vaisala, Vantaa, Finland) at the exit of the reaction cell. Additionally, temperature was measured using another small temperature sensor (Pt 100, 1/3 DIN B, Heraeus, Hanau, Germany) in the reaction cell. All the experiments were conducted at 298 ± 0.5 K. The experiments were conducted under two RHs (72 and 82 %) at 75 ppm SO2 and 75 ppm NO2.
During the reaction, particles were monitored in situ via a glass window on the top of the reaction cell using a Micro-Raman spectrometer (LabRam HR800, HORIBA Jobin Yvon, Kyoto, Japan) to obtain microscopic images and Raman spectra. A 514 nm excitation laser was used, and back-scattering Raman signals were detected. The details of the instrument are described elsewhere (Liu et al., 2008; Zhao et al., 2011). Because the particles were larger than the laser spot in this study (∼ 1.5 µm), confocal Raman mapping was used to measure the spectra at different locations on a particle to obtain the chemical information of the entire particle. The mapping area was rectangular and was slightly larger than the particle, with mapping steps of 1 × 1 µm. Raman spectra in the range of 800–3900 cm−1 were acquired with an exposure time of 1 s for each mapping point. Raman spectra were analyzed using Labspec 5 software (HORIBA Jobin Yvon). Raman peaks were fitted to Gaussian–Lorentzian functions to obtain peak positions and peak areas at different locations on the particle. The peak areas were then added together to obtain the peak area for the entire particle.
Particles of CaCO3 (98 %, Sigma-Aldrich, USA), with average diameters of about 7–10 µm, as specified by the supplier, were used in the experiments. The CaCO3 particles were rhombohedron crystals; X-ray diffraction analysis indicated that they were calcite (Fig. S2). Individual particles were prepared by dripping a dilute CaCO3 suspended solution onto Teflon® FEP film using a pipette and then drying the sample in an oven at 80 ∘C for 10 h.
The amount of CaSO4 as a reaction product was quantified based on Raman peak areas and particle sizes. The details of the method are described in our previous study (Zhao et al., 2018). Briefly, the amount of reaction product CaSO4 formed was determined as a function of time using Raman peak areas. Raman peak areas were converted to the amount of compound formed using a calibration curve obtained from pure CaSO4 particles of different sizes, which were determined according to microscopic images. The reaction rate, i.e., the sulfate production rate, was derived from the amount of sulfate formed as a function of time. The reactive uptake coefficient of SO2 for sulfate formation (γ) was further determined from the reaction rate and collision rate of SO2 on the surface of a single particle.
where R is the gas constant, T is temperature, is the molecular weight of SO2, c is the mean molecular velocity of SO2, As is the surface area of an individual particle, and Z is the collision rate of SO2 on the surface of a particle. indicates the amount of sulfate in the particle phase in moles. The average reaction rate and surface area of particles during the multiphase reaction period were used to derive the reactive uptake coefficient. The period was chosen to start after the induction period when ∼ 10 % of the final sulfate was formed. [SO2] indicates the concentration of SO2 in the gas phase.
The influence of gas-phase diffusion on reactive uptake was evaluated using the resistor model described by Davidovits et al. (2006) and references therein, as well as using the gas-phase diffusion correction factor for a reactive uptake coefficient according to the method described by Pöschl et al. (2007). The reactive uptake of SO2 was found to not be limited by gas-phase diffusion (see details in the Supplement, Sect. S1).
In addition, we conducted experiments of the reaction SO2 with only O2 on both CaCO3 solid particles and internally mixed CaCO3 ∕ Ca(NO3)2 particles (with CaCO3 embedded in Ca(NO3)2 droplets), while keeping other conditions the same as the reaction of SO2 with an O2 ∕ NO2 mixture. These experiments of the multiphase oxidation of SO2 by O2 can help determine the role of NO2 in the reaction of SO2 with an O2 ∕ NO2 mixture.
3.1 Reaction products and changes in particle morphology
Figure 1 shows the Raman spectra of a CaCO3 particle during the multiphase reaction of SO2 with O2 ∕ NO2 ∕ H2O on its surface. The peak at 1087 cm−1 was assigned to the symmetric stretching of carbonate (νs()) (Nakamoto, 1997). During the reaction, the peak at 1087 cm−1 decreased continuously and finally disappeared as new peaks were observed. The peak at 1050 cm−1 was assigned to the symmetric stretching of nitrate (νs()). The peaks at 1010 and 1136 cm−1 were assigned to the symmetric stretching (νs()) and asymmetric stretching (νas()) of sulfate in gypsum (CaSO4 • 2H2O), respectively (Sarma et al., 1998). In addition, after the reaction, a broad envelope in the range of 2800–3800 cm−1 assigned to the stretching of the OH bond in water molecules was observed. Above this envelope, there were two peaks at 3408 and 3497 cm−1, which were assigned to OH bond stretching in crystallization water of CaSO4 • 2H2O (Sarma et al., 1998; Ma et al., 2013).

Figure 1Raman spectra of a CaCO3 particle during the multiphase reaction of SO2 with O2 ∕ NO2 ∕ H2O on the particle. SO2: 75 ppm, NO2: 75 ppm, RH: 72 %, O2: 20 %. The peak intensity of carbonate (1087 cm−1) at 0 and 42 min was divided by 3 for clarity.

Figure 2Microscopic image (i) and Raman mapping image of carbonate (ii), nitrate (iii), and sulfate (iv) on the CaCO3 particle during the multiphase reaction SO2 with O2 ∕ NO2 ∕ H2O on the particle. Panels (a–f) correspond to the reaction times of 0, 20, 41, 76, 117, and 193 min. SO2: 75 ppm, NO2: 75 ppm, RH: 72 %, O2: 20 %. The mapping images of carbonate, nitrate, and sulfate are made using the peak area at 1050, 1010, and 1087 cm−1, respectively. The red, blue, and green colors indicate the peak intensity of carbonate, nitrate, and sulfate, respectively. The dashed lines in panel (iii.f) and (iv.f) indicate the shape of the droplet at the end of the reaction.
During the multiphase reaction with the SO2 ∕ O2 ∕ NO2 ∕ H2O mixture, the CaCO3 particles displayed a remarkable change in morphology. The original CaCO3 particle was a rhombohedron crystal (Fig. 2i.a). As the reaction proceeded, its edges became smoother and later a transparent droplet layer formed, which had a newly formed solid phase embedded in it (Fig. 2i.d). The size of the new solid phase grew during the reaction (Fig. 2i.d–f) and it seemed to contain many micro-crystals. Raman mapping revealed that the new solid phase consisted of CaSO4 • 2H2O (Fig. 2iv), and the surrounding aqueous layer consisted of Ca(NO3)2 (Fig. 2iii).
The particle morphology change shown in Fig. 2 was significantly different from the morphology change in the direct reaction of SO2 with NO2 (Zhao et al., 2018), where the CaCO3 particle was first converted to a spherical Ca(NO3)2 droplet and then needle-shaped CaSO4 crystals formed inside the droplet (Zhao et al., 2018). Moreover, the amount of CaSO4 formed in this study was much higher than that in the direct reaction of SO2 with NO2. The CaSO4 solid particle constituted a significant fraction of the volume of the droplet, while in the direct reaction of SO2 with NO2 the few needle-shaped CaSO4 crystals that formed only constituted a small fraction of the droplet volume (Zhao et al., 2018).
3.2 Reaction process
During the reaction, the amounts of carbonate, nitrate, and sulfate were determined as a function of time, as shown in Fig. 3. At the beginning of the reaction, the amount of carbonate decreased slowly, while the amount of nitrate and sulfate increased slowly. After a period of induction of around 50 min, the reaction accelerated significantly, leading to a rapid consumption of carbonate and production of nitrate and sulfate. The decrease in the amount of carbonate and the increase in the amount of nitrate was because carbonate reacted continuously with NO2 and H2O, forming Ca(NO3)2. The detailed mechanisms of the multiphase reaction of carbonate with NO2 and H2O were discussed in our previous studies (Li et al., 2010; Zhao et al., 2018). The mechanism of sulfate formation is discussed in detail in Sect. 3.4 of the present study. Finally, the carbonate was completely consumed, and the amounts of nitrate and sulfate leveled off.
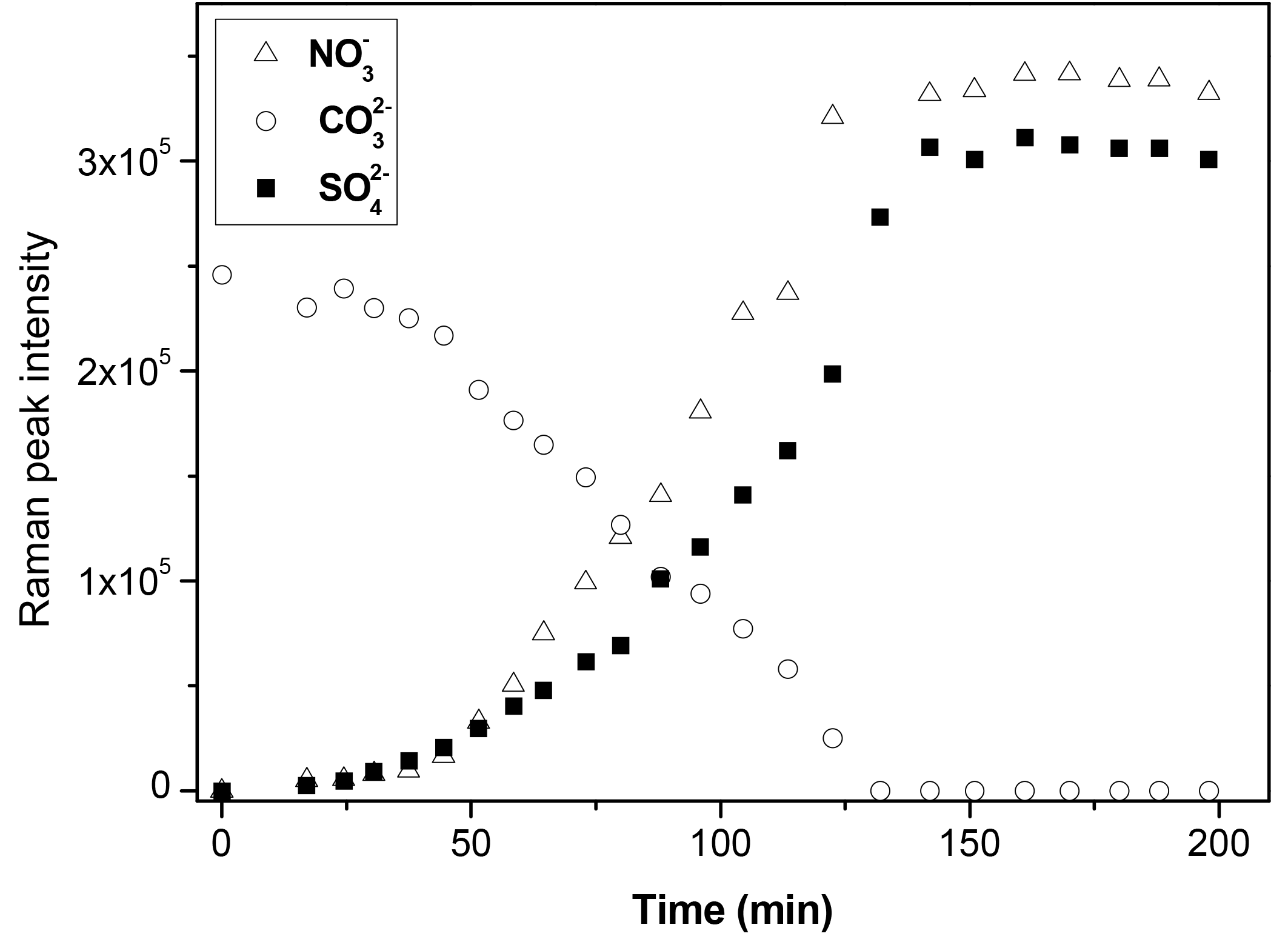
Figure 3Time series of the Raman peak intensity of , , and during the reaction of SO2 with O2 ∕ NO2 ∕ H2O on CaCO3 particles. SO2: 75 ppm, NO2: 75 ppm, RH: 72%, O2: 20 %. The intensity of , , and show the peak area at 1050, 1010, and 1087 cm−1, respectively, in Raman spectra obtained by Raman mapping.
Figure 3 shows that nitrate and sulfate were formed simultaneously during the reaction. This contrasts with the observations made during the direct reaction of SO2 with NO2, where nitrate was formed first, and sulfate was essentially formed after the complete conversion of CaCO3 particles to Ca(NO3)2 droplets (Zhao et al., 2018). Moreover, the time taken for carbonate to be completely consumed was longer in this study than in the direct reaction of SO2 with NO2 (∼ 120 vs. ∼ 40 min) when other conditions were kept the same (Zhao et al., 2018).
3.3 Reactive uptake coefficient of SO2
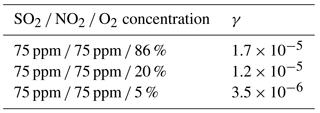
The reactive uptake coefficients of SO2 for sulfate formation (γ) in the reaction of SO2 with the O2 ∕ NO2 ∕ H2O ∕ N2 mixture on CaCO3 with various O2 concentrations are shown in Table 1. The value of γ for the reaction of SO2 with O2 ∕ NO2 at three O2 concentrations (5, 20, and 86 %) was in the range of (0.35–1.7) × 10−5, and was 1.2 × 10−5 in synthetic air. This latter value was 2–3 orders of magnitude higher than that for the reaction of SO2 directly with NO2 under similar conditions (Zhao et al., 2018). When other conditions were kept constant, γ increased with the O2 concentration. This indicates that O2 played a key role in enhancing the oxidation rate of SO2.
Table 1Reactive uptake coefficient of SO2 for sulfate formation at 82 % RH and at different O2 concentrations.
The role of O2 in enhancing the reactive uptake of SO2 reported here is consistent with the findings in some previous studies. For example, the data of Littlejohn et al. (1993) showed that the sulfite oxidation rate increases with the O2 concentration (0–5 % by volume). Shen and Rochelle (1998) also found that in the presence of O2, the aqueous sulfite oxidation rate is enhanced. By investigating the oxidation of SO2 by NO2 in monodispersed water droplets growing on carbon nuclei, Santachiara et al. (1990) found that the sulfate formation rate with 2 % O2 is much higher than that without O2. However, our findings, as well as those in the studies referred to above, are in contrast to those reported by Lee and Schwartz (1983), who found that changing from N2 to air as a carrier gas only increases the bisulfite oxidation rate by no more than 10 %. The difference between our study and Lee and Schwartz (1983) could be due to the difference in O2 diffusion from the gas to the condensed phase and the different mechanisms between the multiphase reaction on particles and the aqueous reaction.
Only few studies have reported the S(IV) oxidation rate in the reaction of S(IV) with O2 ∕ NO2 mixtures (Turšič et al., 2001; Littlejohn et al., 1993). However, due to the limiting step by the aqueous-phase mass transfer, it is difficult to quantitatively compare the reaction rates in those studies with the uptake coefficient in our study and the rate constants determined by Lee and Schwartz (1983) and Clifton et al. (1988). For example, a rate constant of 2.4 × 103 mol−1 L s−1 (at pH 3) can be derived from the results of Turšič et al. (2001), which is much lower than the values reported by Lee and Schwartz (1983) and Clifton et al. (1988). This can be attributed to the limiting step by the aqueous-phase mass transfer because the characteristic mixing time in the aqueous phase in Turšič et al. (2001) was likely much longer than that of Lee and Schwartz (1983) (1.7–5.3 s), according to the HSO concentration time series reported by Turšič et al. (2001).
It is important to note that the concentrations of NO2 and SO2 used in this study are much higher than those in the ambient atmosphere. High concentrations of reactant gases are often used in laboratory studies in order to simulate the ambient reactions at the timescale of days or weeks and to obtain high signal-to-noise ratios for detecting products within minutes or hours. In the ambient atmosphere, the reactive uptake coefficient of SO2 should be lower than that in this study due to the lower NO2 concentrations when other conditions are comparable, and the chemical and physical processes observed in this study, such as changes in particle composition, phase, hygroscopicity, and pH, should be much slower due to the lower concentrations of NO2 and SO2.
3.4 Reaction mechanism
In the multiphase reaction of SO2 with O2 ∕ NO2 ∕ H2O on CaCO3 particles, we found that CaCO3 reacted with NO2 and H2O and produced Ca(NO3)2, which deliquesced, forming liquid water, and provided a site for the aqueous oxidation of SO2. This process is similar to the direct reaction of SO2 with NO2 on CaCO3 particles. The details of this part of the reaction mechanism were discussed in our previous study (Zhao et al., 2018).
Once the aqueous phase was formed, SO2 could undergo multiphase reactions with O2 ∕ NO2. The mechanism of the direct aqueous reaction of S(IV) with NO2 in the absence of O2 is complex. Previous studies have proposed two different mechanisms for the reaction. One involves radical formation (Littlejohn et al., 1993; Shen and Rochelle, 1998; Turšič et al., 2001) (referred to as the “free-radical chain” mechanism), while the other involves the formation of NO2–S(IV) complexes (Clifton et al., 1988), but no radical formation (referred to as the “NO2–S(IV) complex” mechanism).
Table 2Summary of the mechanism of the reaction S(IV) with O2 ∕ NO2.

According to the NO2–S(IV) complex mechanism, the presence of O2 should not affect the SO2 oxidation rate; however, in this study, a substantial enhancement in the SO2 oxidation rate was observed in the presence of O2 compared with that in the absence of O2. Therefore, the NO2–S(IV) complex mechanism was less likely to have been important in this study.
In the free-radical chain mechanism, the radical is proposed to be formed (Reaction R8, Table 2), which is based on the observation of formation, with , known to be the combination reaction product of (Eriksen, 1974; Hayon et al., 1972; Deister and Warneck, 1990; Brandt et al., 1994; Waygood and McElroy, 1992). In addition to and , was detected with an appreciable yield using Raman spectroscopy, following the reaction of NO2 with aqueous sulfite (Littlejohn et al., 1993). was also observed in the aqueous oxidation of bisulfite in an N2-saturated solution in the presence of Fe(III) using ion interaction chromatography (Podkrajšek et al., 2002). The radical can react via two pathways, forming either or (Reactions R9–R11, Table 2). Reactions R9–R11 have been well established in studies of S(IV) oxidation by other pathways, including OH oxidation, photo-oxidation, and transition metal catalyzed oxidation (Eriksen, 1974; Hayon et al., 1972; Deister and Warneck, 1990; Brandt et al., 1994; Brandt and Vaneldik, 1995; Waygood and McElroy, 1992). In addition, although previous studies have not reported the direct observation of the radical in the aqueous reaction of S(IV) with NO2, was observed in the reaction of with in an acidic buffer solution (pH = 4.0) using electron spin resonance (Shi, 1994). Because is formed in the aqueous reaction of SO2 with NO2, and as the combination reaction product of is observed (Littlejohn et al., 1993), formation is plausible.
In the presence of O2, the radical can react rapidly with O2, forming the radical (Reaction R12, Table 2). Following this reaction, a number of chain reactions can occur to ultimately form sulfate (Littlejohn et al., 1993; Seinfeld and Pandis, 2006; Shen and Rochelle, 1998) (Reactions R13–R16, Table 2). Littlejohn et al. (1993) observed that the amount of relative to formed in the aqueous reaction of NO2 with sulfite decreases in the presence of O2 compared with the reaction in the absence of O2. At low NO2 concentrations (< 5 ppm), is undetectable in the presence of O2. This indicates that O2 suppresses the reaction pathway of formation (Reaction R9, Table 2). Because the radical can react rapidly with O2, forming the radical, and would therefore be consumed, the suppression of formation can be attributed to the reaction of with O2 (Reaction R12, Table 2). Reactions (R12)–(R16) have been well established by studies of the oxidation of S(IV) by OH or photo-oxidation, and all the radicals have been observed (Hayon et al., 1972; Huie et al., 1989; Huie and Neta, 1987; Chameides and Davis, 1982; Seinfeld and Pandis, 2006).
The free-radical chain mechanism is consistent with the findings of this study and is therefore more plausible. The enhancement of the SO2 oxidation rate in the reaction of SO2 with O2 ∕ NO2 ∕ H2O on CaCO3 particles compared with that in the direct reaction of SO2 with NO2 ∕ H2O was attributed to O2. Although during the reaction in the absence of O2 – i.e., the direct oxidation of SO2 by NO2 – the radical can be formed (Reaction R7), the reaction chain cannot propagate (Reactions R12–R16). Therefore, the S(IV) oxidation rate and the reactive uptake coefficient of SO2 were much lower than those in the presence of O2. According to the difference between the reactive uptake coefficient in this study and in the direct reaction of SO2 with NO2 (Zhao et al., 2018), the sulfate production rate via chain reactions due to the presence of O2 (20 %) was 2–3 orders of magnitude faster than the direct oxidation of SO2 by NO2. This indicates that sulfate production in the reaction of SO2 with O2 ∕ NO2 was largely due to O2 oxidation via the chain reaction pathway, i.e., “autoxidation” of S(IV), rather than the direct oxidation of SO2 by NO2, and thus O2 was the main oxidant of SO2.
In addition to the two mechanisms above, Spindler et al. (2003) proposed a reaction mechanism involving first NO2–S(IV) complex formation and subsequent radical formation (Reactions R3, R7). NO2–S(IV) complexes may establish an equilibrium with in contrast to the direct formation of SO via the reaction of NO2 with SO2. Higher concentration of O2 favors the conversion of to and thus can reduce the concentration, shifting the equilibrium to the product side and promoting the overall S(IV) oxidation. O2 can act in a similar way as in the free-radical chain mechanism. Admittedly, we cannot rule out the possibility of a NO2–S(IV) complex formation. But such a mechanism can still be classified as the free-radical chain mechanism since the S(IV) oxidation still proceeds via the radical chain reactions. Although the direct oxidation of SO2 by NO2 only accounted for a very small fraction of sulfate formation, NO2 played an important role in the SO2 oxidation by initiating the chain reactions via the production of the radical (Reaction R7). In the experiment without NO2, but with other reaction conditions the same, we were unable to detect sulfate after 5 h of reaction. This indicates that O2 by itself cannot initiate the chain reactions (although it favors chain propagation), and that the oxidation of SO2 by O2 was slow. The effect on the SO2 oxidation rate when both NO2 and O2 were present was much higher than the sum of the effect of NO2 and O2. We refer to this effect as the synergy of NO2 and O2, which resulted in the fast oxidation of SO2 to form sulfate in this study. This effect is similar to a “ternary” reaction found with the reaction of NO2–particles–H2O or SO2–particles–O3 (Zhu et al., 2011), where the reaction rate can be much faster than the sum of the reaction rates for the reaction of the second and third reactant with the first reactant. In addition to acting as the initiator of chain reactions, NO2 also contributed to the formation of the aqueous phase through the reaction with CaCO3, forming Ca(NO3)2 as discussed above, which provided a site for S(IV) oxidation.
Based on the discussion above, we summarize the reaction mechanism that occurred in this study in Table 2. The reactions are classified as chain initiation, chain propagation, and chain termination. The dominant S(IV) species depends on pH. Due to the fast dissociations of SO2 • H2O and , reactions consuming one of these S(IV) species will result in instantaneous re-establishment of the equilibria between them (Seinfeld and Pandis, 2006). In this study, the pH of the aqueous layer of Ca(NO3)2 may change dynamically with time during the reaction and may not be completely homogeneous within the aqueous droplet. The pH values could vary between ∼ 3 and ∼ 7.6. In the surface of the aqueous layer, pH was mainly determined by the gas–aqueous equilibrium of SO2, and was estimated to be ∼ 3. In the vicinity of the CaCO3 core, pH was mainly determined by the hydrolysis of carbonate and was estimated to be ∼ 7.6. It is likely that both HSO and SO were present, and the dominant species depended on the reaction time and location within the aqueous droplet. Nevertheless, to make the reaction mechanism clearer, HSO was used in the reaction equations. Similar reaction equations are also applicable to SO because of the fast dissociations of SO2 • H2O and . Overall, the reaction can be written as follows, which clearly shows that O2 was the main oxidant for sulfate formation:
where n ≫ 1. Once sulfuric acid was formed, it reacted with CaCO3, forming CaSO4:
As mentioned above, compared with the direct reaction of SO2 with NO2, CaCO3 was consumed more slowly in the reaction with O2 ∕ NO2. There were two possible reasons for this. First, the CaSO4 • 2H2O formed in the reaction could cover the CaCO3 surface and partly suppress the diffusion of aqueous ions, such as protons, and also limit the contact of reactants with the surface of the CaCO3 particles, thus reducing the CaCO3 consumption rate. Second, compared with the direct reaction of SO2 with NO2, a much higher fraction of CaCO3 was converted to CaSO4 • 2H2O instead of Ca(NO3)2 due to the fast production of CaSO4 • 2H2O. Therefore, the volume of a Ca(NO3)2 droplet was much smaller than that in the direct reaction of SO2 with NO2 for a given CaCO3 particle. Because the uptake rate of NO2 was proportional to the droplet surface area and the NO2 hydrolysis rate was proportional to the droplet volume, the rate of nitric acid production from NO2 hydrolysis and its reaction rate with CaCO3 were reduced. Therefore, the CaCO3 particles were consumed more slowly in the reaction with O2 ∕ NO2.
We investigated the multiphase reaction of SO2 with O2 ∕ NO2 ∕ H2O on CaCO3 particles. The reaction converted CaCO3 particles to Ca(NO3)2 droplets, in which CaSO4 • 2H2O was embedded and accounted for a significant fraction of the droplet volume by the end of the reaction. The Ca(NO3)2 droplet formed by the reaction of CaCO3 with NO2 provided a site for the multiphase oxidation of SO2. Generally, nitrate and sulfate were formed simultaneously. The reactive uptake coefficient of SO2 for sulfate formation in the reaction of SO2 with NO2 ∕ H2O in synthetic air was determined to be around 10−5. Compared with the reaction of SO2 with NO2 on a CaCO3 particle in the absence of O2, i.e., the direct oxidation of SO2 by NO2 in N2, the sulfate production rate in the reaction of SO2 with O2 ∕ NO2 was enhanced by 2–3 orders of magnitude. According to the findings of this study and the existing literature, SO2 oxidation likely proceeded via a free-radical chain reaction mechanism. O2 was the main oxidant of SO2, and NO2 mainly acted as an initiator of the chain reactions. The synergy of NO2 and O2 resulted in the fast oxidation of SO2. The absence of either NO2 or O2 led to much slower SO2 oxidation.
Using a method developed in our previous study (Zhao et al., 2018), we assessed the importance of the multiphase oxidation of SO2 by O2 in the presence of NO2 by estimating the lifetime of SO2 due to multiphase reactions and the lifetime due to the gas-phase reaction (with the OH radical). The lifetime of SO2 due to the multiphase reaction of SO2 with O2 ∕ NO2 was estimated to be around 20 days using the reactive uptake coefficient of SO2 (1.2 × 10−5) and the typical particle surface area concentration for mineral aerosols in winter in Beijing (6.3 × 10−6 cm2 cm−3) (Huang et al., 2015). This lifetime is comparable to the lifetime of SO2 due to the gas-phase reaction with OH, which is ∼ 12 days, assuming that the daytime OH concentration is 1 × 106 molecules cm−3 (Lelieveld et al., 2016; Prinn et al., 2005). Therefore, we conclude that the multiphase oxidation of SO2 by O2 in the presence of NO2 is likely to be an important source of sulfate and a sink of SO2 in the ambient atmosphere, and can play a significant role in the sulfate formation during severe haze episodes, such as those that frequently occur in China. During haze episodes, there are high concentrations of SO2 and NO2 and relative humidity is often high (Zhang et al., 2014; Zheng et al., 2015b; Wang et al., 2016). Under these conditions, the multiphase oxidation of SO2 by O2 in the presence of NO2 could proceed rapidly, forming sulfate. The enhanced sulfate concentration due to multiphase reactions and resulting aerosol water content can further promote the multiphase oxidation of SO2. The reaction thus proceeds in a self-accelerating way. Therefore, it can contribute significantly to sulfate formation during haze episodes, which could explain the discrepancies between the observed and modeled sulfate concentrations (Cheng et al., 2016; Gao et al., 2016; Wang et al., 2016; Zheng et al., 2015a).
In addition, elucidating the mechanism of the multiphase reaction of SO2 with O2 ∕ NO2 ∕ H2O in the atmosphere is important for the other atmospheric implications of the reaction besides sulfate formation. According to the reaction mechanism, the direct oxidation of SO2 by NO2 forms sulfate and nitrite, with a stoichiometry of 1 : 1, and nitrite can further form HONO under acidic conditions. The HONO could then evaporate into the atmosphere, where it would be an important source of OH radicals. If NO2 were the main oxidant of SO2 in the multiphase reaction, the reaction would form one HONO molecule for every sulfate molecule formed. Thus, the oxidation of SO2 by NO2 can simultaneously be an important source of HONO and OH radicals, and SO2 oxidation would be strongly coupled with reactive nitrogen chemistry. However, according to the mechanism of this study, NO2 only acted as an initiator of the chain reactions in SO2 oxidation and essentially all the SO2 was oxidized by O2. Therefore, the amount of HONO formation per sulfate molecule formed was trivial. The oxidation of SO2 by O2 ∕ NO2 is expected to be neither an important source of HONO and OH in the atmosphere nor to have a significant influence on reactive nitrogen chemistry.
In this study, we investigated the reaction of SO2 with O2 in the presence of NO2 at three O2 concentrations. The influence of the O2 concentration was shown to be significant. Future experimental results with more O2 concentration levels would provide more insight into the reaction mechanism and process.
In addition, in the ambient atmosphere, the internal mixing of organics with S(IV) in particles may influence the S(IV) oxidation rate by O2 in the presence of NO2. When organics are abundant in particles, for example during haze episodes in China, they can react with and thus scavenge radical anion carriers such as and (Herrmann, 2003; Herrmann et al., 2015; Huie, 1995). Therefore, the presence of internally mixed organics can reduce the effectivity of the potential radical reaction chain and of S(IV) oxidation, which can undermine the importance of the oxidation by O2 in the presence of NO2 in the overall S(IV) oxidation.
The data in the figures in both the main text and the Supplement are available upon request to the corresponding author (tzhu@pku.edu.cn).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-6679-2018-supplement.
The authors declare that they have no conflict of interest.
This work was supported by the Natural Science Foundation Committee of China
(41421064, 21190051, 40490265, 91544000) and the Ministry of Science and
Technology (grant no. 2002CB410802).
Edited
by: Markus Ammann
Reviewed by: three anonymous referees
Brandt, C. and Vaneldik, R.: Transition metal-catalyzed oxidation of sulfur (IV) oxides. Atmospheric-relevant processes and mechanisms, Chem. Rev., 95, 119–190, https://doi.org/10.1021/cr00033a006, 1995.
Brandt, C., Fabian, I., and Vaneldik, R.: Kinetics and mechanism of the iron(III)-catalyzed autoxidation of sulfur(IV) oxides in aqueous-solution – evidence for the redox cycling of iron in the presence of oxygen and modeling of the overall reaction-mechanism, Inorg. Chem., 33, 687–701, https://doi.org/10.1021/ic00082a012, 1994.
Chameides, W. L. and Davis, D. D.: The free-radical chemistry of cloud droplets and its impact upon the composition of rain, J. Geophys. Res.-Oceans, 87, 4863–4877, https://doi.org/10.1029/JC087iC07p04863, 1982.
Cheng, Y. F., Zheng, G. J., Wei, C., Mu, Q., Zheng, B., Wang, Z. B., Gao, M., Zhang, Q., He, K. B., Carmichael, G., Poschl, U., and Su, H.: Reactive nitrogen chemistry in aerosol water as a source of sulfate during haze events in China, Sci. Adv., 2, e1601530, https://doi.org/10.1126/sciadv.1601530, 2016.
Clifton, C. L., Altstein, N., and Huie, R. E.: Rate-constant for the reaction of NO2 with sulfur(IV) over the pH range 5.3–13, Environ. Sci. Technol., 22, 586–589, https://doi.org/10.1021/es00170a018, 1988.
Davidovits, P., Kolb, C. E., Williams, L. R., Jayne, J. T., and Worsnop, D. R.: Mass accommodation and chemical reactions at gas-liquid interfaces, Chem. Rev., 106, 1323–1354, https://doi.org/10.1021/cr040366k, 2006.
Deister, U. and Warneck, P.: Photooxidation of sulfite () in aqueous solution, J. Phys. Chem., 94, 2191–2198, https://doi.org/10.1021/j100368a084, 1990.
Eriksen, T. E.: pH Effects on the pulse radiolysis of deoxygenated aqueous solutions of sulphur dioxide, Journal of the Chemical Society, Faraday Transactions 1: Physical Chemistry in Condensed Phases, 70, 208–215, https://doi.org/10.1039/f19747000208, 1974.
Gao, M., Carmichael, G. R., Wang, Y., Ji, D., Liu, Z., and Wang, Z.: Improving simulations of sulfate aerosols during winter haze over Northern China: the impacts of heterogeneous oxidation by NO2, Front. Environ. Sci. En., 10, 16, https://doi.org/10.1007/s11783-016-0878-2, 2016.
Guo, H., Weber, R. J., and Nenes, A.: High levels of ammonia do not raise fine particle pH sufficiently to yield nitrogen oxide-dominated sulfate production, Sci. Rep., 7, 12109, https://doi.org/10.1038/s41598-017-11704-0, 2017.
Hayon, E., Treinin, A., and Wilf, J.: Electronic spectra, photochemistry, and autoxidation mechanism of the sulfite-bisulfite-pyrosulfite systems. SO2-, SO3-, SO4-, and SO5-radicals, J. Am. Chem. Soc. , 94, 47–57, https://doi.org/10.1021/ja00756a009, 1972.
He, H., Wang, Y., Ma, Q., Ma, J., Chu, B., Ji, D., Tang, G., Liu, C., Zhang, H., and Hao, J.: Mineral dust and NOx promote the conversion of SO2 to sulfate in heavy pollution days, Sci. Rep., 4, 4172, https://doi.org/10.1038/srep04172, 2014.
Herrmann, H.: Kinetics of aqueous phase reactions relevant for atmospheric chemistry, Chem. Rev., 103, 4691–4716, https://doi.org/10.1021/cr020658q, 2003.
Herrmann, H., Schaefer, T., Tilgner, A., Styler, S. A., Weller, C., Teich, M., and Otto, T.: Tropospheric Aqueous-Phase Chemistry: Kinetics, Mechanisms, and Its Coupling to a Changing Gas Phase, Chem. Rev., 115, 4259–4334, https://doi.org/10.1021/cr500447k, 2015.
Huang, L., Zhao, Y., Li, H., and Chen, Z.: Kinetics of Heterogeneous Reaction of Sulfur Dioxide on Authentic Mineral Dust: Effects of Relative Humidity and Hydrogen Peroxide, Environ. Sci. Technol., 49, 10797–10805, https://doi.org/10.1021/acs.est.5b03930, 2015.
Huie, R. E. and Neta, P.: Kinetics of one-electron transfer-reactions involving ClO2 and NO2, J. Phys. Chem. , 90, 1193–1198, https://doi.org/10.1021/j100278a046, 1986.
Huie, R. E. and Neta, P.: Rate constants for some oxidations of S(IV) by radicals in aqueous-solutions, Atmos. Environ., 21, 1743–1747, https://doi.org/10.1016/0004-6981(87)90113-2, 1987.
Huie, R. E., Clifton, C. L., and Altstein, N.: A pulse radiolysis and flash photolysis study of the radicals SO2, SO3, SO4 and SO5, Radiat. Phys. Chem., 33, 361–370, 1989.
Huie, R. E.: Free radical chemistry of the atmospheric aqueous phase, in: Progress and Problems in Atmospheric Chemistry, WORLD SCIENTIFIC, Singapore, 374–419, 1995.
Lee, Y.-N. and Schwartz, S. E.: Kinetics of oxidation of aqueous sulfur (IV) by nitrogen dioxide, in: Precipitation Scavenging, Dry Deposition and Resuspension, edited by: Pruppacher, H. R., Semonin, R. G., and Slinn, W. G. N., Elsevier, New York, 453–466, 1983.
Lelieveld, J., Gromov, S., Pozzer, A., and Taraborrelli, D.: Global tropospheric hydroxyl distribution, budget and reactivity, Atmos. Chem. Phys., 16, 12477–12493, https://doi.org/10.5194/acp-16-12477-2016, 2016.
Li, H. J., Zhu, T., Zhao, D. F., Zhang, Z. F., and Chen, Z. M.: Kinetics and mechanisms of heterogeneous reaction of NO2 on CaCO3 surfaces under dry and wet conditions, Atmos. Chem. Phys., 10, 463–474, https://doi.org/10.5194/acp-10-463-2010, 2010.
Littlejohn, D., Wang, Y. Z., and Chang, S. G.: Oxidation of aqueous sulfite ion by nitrogen-dioxide, Environ. Sci. Technol., 27, 2162–2167, https://doi.org/10.1021/es00047a024, 1993.
Liu, M. X., Song, Y., Zhou, T., Xu, Z. Y., Yan, C. Q., Zheng, M., Wu, Z. J., Hu, M., Wu, Y. S., and Zhu, T.: Fine particle pH during severe haze episodes in northern China, Geophys. Res. Lett., 44, 5213–5221, https://doi.org/10.1002/2017gl073210, 2017.
Liu, Y. J., Zhu, T., Zhao, D. F., and Zhang, Z. F.: Investigation of the hygroscopic properties of Ca(NO3)2 and internally mixed Ca(NO3)2 ∕ CaCO3 particles by micro-Raman spectrometry, Atmos. Chem. Phys., 8, 7205–7215, https://doi.org/10.5194/acp-8-7205-2008, 2008.
Ma, Q., He, H., Liu, Y., Liu, C., and Grassian, V. H.: Heterogeneous and multiphase formation pathways of gypsum in the atmosphere, Phys. Chem. Chem. Phys., 15, 19196-19204, 10.1039/c3cp53424c, 2013.
Nakamoto, K.: Infrared and Raman Spectra of Inorganic and Coordination Compounds Part A, John Wiley &Sons, New York, 221–247, 1997.
Nash, T.: Effect of nitrogen-dioxide and of some transition-metals on the oxidation of dilute bisulfite solutions, Atmos. Environ., 13, 1149–1154, https://doi.org/10.1016/0004-6981(79)90038-6, 1979.
Podkrajšek, B., Grgić, I., and Turšič, J.: Determination of sulfur oxides formed during the S(IV) oxidation in the presence of iron, Chemosphere, 49, 271–277, https://doi.org/10.1016/S0045-6535(02)00324-7, 2002.
Pöschl, U., Rudich, Y., and Ammann, M.: Kinetic model framework for aerosol and cloud surface chemistry and gas-particle interactions – Part 1: General equations, parameters, and terminology, Atmos. Chem. Phys., 7, 5989–6023, https://doi.org/10.5194/acp-7-5989-2007, 2007.
Prinn, R. G., Huang, J., Weiss, R. F., Cunnold, D. M., Fraser, P. J., Simmonds, P. G., McCulloch, A., Harth, C., Reimann, S., Salameh, P., O'Doherty, S., Wang, R. H. J., Porter, L. W., Miller, B. R., and Krummel, P. B.: Evidence for variability of atmospheric hydroxyl radicals over the past quarter century, Geophys. Res. Lett., 32, L07809, https://doi.org/10.1029/2004gl022228, 2005.
Santachiara, G., Prodi, F., and Vivarelli, F.: SO2 oxidation in monodisperse droplets grown on carbon nuclei in presence of NO2, J. Aerosol Sci., 21, S221–S224, https://doi.org/10.1016/0021-8502(90)90224-l, 1990.
Santachiara, G., Prodi, F., and Vivarelli, F.: Further experiments on SO2 oxidation rate in monodisperse droplets grown on carbon nuclei in presence of O-2 and NO2, J. Aerosol Sci., 24, 683–685, https://doi.org/10.1016/0021-8502(93)90024-4, 1993.
Sarma, L. P., Prasad, P. S. R., and Ravikumar, N.: Raman spectroscopic study of phase transitions in natural gypsum, J. Raman Spectrosc., 29, 851–856, https://doi.org/10.1002/(sici)1097-4555(199809)29:9<851::aid-jrs313>3.0.co;2-s, 1998.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: from air pollution to climate change, 2nd Edn., John Wiley &Sons. Inc., 2006.
Shen, C. H. and Rochelle, G. T.: Nitrogen Dioxide Absorption and Sulfite Oxidation in Aqueous Sulfite, Environ. Sci. Technol., 32, 1994–2003, https://doi.org/10.1021/es970466q, 1998.
Shi, X. L.: Generation of and OH radicals in reactions with inorganic environmental-pollutants and its implications to SO toxicity, J. Inorg. Biochem., 56, 155–165, https://doi.org/10.1016/0162-0134(94)85002-x, 1994.
Spindler, G., Hesper, J., Bruggemann, E., Dubois, R., Muller, T., and Herrmann, H.: Wet annular denuder measurements of nitrous acid: laboratory study of the artefact reaction of NO2 with S(IV) in aqueous solution and comparison with field measurements, Atmos. Environ., 37, 2643–2662, https://doi.org/10.1016/s1352-2310(03)00209-7, 2003.
Turšič, J., Grgić, I., and Bizjak, M.: Influence of NO2 and dissolved iron on the S(IV) oxidation in synthetic aqueous solution, Atmos. Environ., 35, 97–104, https://doi.org/10.1016/S1352-2310(00)00283-1, 2001.
Wang, G., Zhang, R., Gomez, M. E., Yang, L., Levy Zamora, M., Hu, M., Lin, Y., Peng, J., Guo, S., Meng, J., Li, J., Cheng, C., Hu, T., Ren, Y., Wang, Y., Gao, J., Cao, J., An, Z., Zhou, W., Li, G., Wang, J., Tian, P., Marrero-Ortiz, W., Secrest, J., Du, Z., Zheng, J., Shang, D., Zeng, L., Shao, M., Wang, W., Huang, Y., Wang, Y., Zhu, Y., Li, Y., Hu, J., Pan, B., Cai, L., Cheng, Y., Ji, Y., Zhang, F., Rosenfeld, D., Liss, P. S., Duce, R. A., Kolb, C. E., and Molina, M. J.: Persistent sulfate formation from London Fog to Chinese haze, P. Natl. Acad. Sci. USA, 113, 13630–13635, https://doi.org/10.1073/pnas.1616540113, 2016.
Waygood, S. J. and McElroy, W. J.: Spectroscopy and decay kinetics of the sulfite radical anion in aqueous solution, J. Chem. Soc.-Faraday Trans., 88, 1525–1530, https://doi.org/10.1039/ft9928801525, 1992.
Xue, J., Yuan, Z. B., Griffith, S. M., Yu, X., Lau, A. K. H., and Yu, J. Z.: Sulfate Formation Enhanced by a Cocktail of High NOx, SO2, Particulate Matter, and Droplet pH during Haze-Fog Events in Megacities in China: An Observation-Based Modeling Investigation, Environ. Sci. Technol., 50, 7325–7334, https://doi.org/10.1021/acs.est.6b00768, 2016.
Zhang, J. K., Sun, Y., Liu, Z. R., Ji, D. S., Hu, B., Liu, Q., and Wang, Y. S.: Characterization of submicron aerosols during a month of serious pollution in Beijing, 2013, Atmos. Chem. Phys., 14, 2887–2903, https://doi.org/10.5194/acp-14-2887-2014, 2014.
Zhao, D., Song, X., Zhu, T., Zhang, Z., Liu, Y., and Shang, J.: Multiphase oxidation of SO2 by NO2 on CaCO3 particles, Atmos. Chem. Phys., 18, 2481–2493, https://doi.org/10.5194/acp-18-2481-2018, 2018.
Zhao, D. F., Zhu, T., Chen, Q., Liu, Y. J., and Zhang, Z. F.: Raman micro-spectrometry as a technique for investigating heterogeneous reactions on individual atmospheric particles, Sci. China Chem., 54, 154–160, https://doi.org/10.1007/s11426-010-4182-x, 2011.
Zheng, B., Zhang, Q., Zhang, Y., He, K. B., Wang, K., Zheng, G. J., Duan, F. K., Ma, Y. L., and Kimoto, T.: Heterogeneous chemistry: a mechanism missing in current models to explain secondary inorganic aerosol formation during the January 2013 haze episode in North China, Atmos. Chem. Phys., 15, 2031–2049, https://doi.org/10.5194/acp-15-2031-2015, 2015a.
Zheng, G. J., Duan, F. K., Su, H., Ma, Y. L., Cheng, Y., Zheng, B., Zhang, Q., Huang, T., Kimoto, T., Chang, D., Pöschl, U., Cheng, Y. F., and He, K. B.: Exploring the severe winter haze in Beijing: the impact of synoptic weather, regional transport and heterogeneous reactions, Atmos. Chem. Phys., 15, 2969–2983, https://doi.org/10.5194/acp-15-2969-2015, 2015b.
Zhu, T., Shang, J., and Zhao, D. F.: The roles of heterogeneous chemical processes in the formation of an air pollution complex and gray haze, Sci. China Chem., 54, 145–153, https://doi.org/10.1007/s11426-010-4181-y, 2011.