the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 09 May 2018
| 09 May 2018
Mass spectrometric measurement of hydrogen isotope fractionation for the reactions of chloromethane with OH and Cl
Frank Keppler
Enno Bahlmann
Markus Greule
Heinz Friedrich Schöler
Julian Wittmer
Cornelius Zetzsch
Chloromethane (CH3Cl) is an important provider of chlorine to the stratosphere but detailed knowledge of its budget is missing. Stable isotope analysis is a potentially powerful tool to constrain CH3Cl flux estimates. The largest degree of isotope fractionation is expected to occur for deuterium in CH3Cl in the hydrogen abstraction reactions with its main sink reactant tropospheric OH and its minor sink reactant Cl atoms. We determined the isotope fractionation by stable hydrogen isotope analysis of the fraction of CH3Cl remaining after reaction with hydroxyl and chlorine radicals in a 3.5 m3 Teflon smog chamber at 293 ± 1 K. We measured the stable hydrogen isotope values of the unreacted CH3Cl using compound-specific thermal conversion isotope ratio mass spectrometry. The isotope fractionations of CH3Cl for the reactions with hydroxyl and chlorine radicals were found to be and ‰, respectively. For comparison, we performed similar experiments using methane (CH4) as the target compound with OH and obtained a fractionation constant of ‰ which is in good agreement with values previously reported. The observed large kinetic isotope effects are helpful when employing isotopic analyses of CH3Cl in the atmosphere to improve our knowledge of its atmospheric budget.
- Article
(745 KB) -
Supplement
(501 KB) - BibTeX
- EndNote





Chloromethane (often called methyl chloride) is the most abundant chlorine-containing trace gas in the Earth's atmosphere, currently with a global mean mixing ratio of ∼ 540 ± 5 parts per trillion by volume (pptv) and an atmospheric lifetime of 1.0–1.2 years (Carpenter et al., 2014). The global emissions of CH3Cl have been estimated to be in the range of 4 to 5 Tg yr−1 (1 Tg = 1012 g) stemming from predominantly natural but also anthropogenic sources (Montzka and Fraser, 2003; WMO, 2011; Carpenter et al., 2014). However, current estimates of the CH3Cl global budget and the apportionment between sources and sinks are still highly uncertain. Known natural sources of CH3Cl include tropical plants (Yokouchi et al., 2002, 2007; Umezawa et al., 2015), wood-rotting fungi (Harper, 1985), oceans (Moore et al., 1996; Kolusu et al., 2017), plants of salt marshes (Rhew et al., 2000, 2003), aerated and flooded soil (Redeker et al., 2000; Keppler et al., 2000), senescent leaves and leaf litter (Hamilton et al., 2003; Derendorp et al., 2011) and wildfires. Anthropogenic CH3Cl release to the atmosphere comes from the combustion of coal and biomass with minor emissions from cattle (Williams et al., 1999) and humans (Keppler et al., 2017). In addition, it has been reported that emissions from industrial sources, particularly in China, might be much higher than previously assumed (Li et al., 2017).
The dominant sink for atmospheric CH3Cl results from the reaction with photochemically produced hydroxyl radicals (OH), currently estimated at about 2.8 Tg yr−1 (Carpenter et al., 2014). Furthermore, in the marine boundary layer the reaction of CH3Cl with chlorine radicals (Cl) represents another sink estimated to account for up to 0.4 Tg yr−1 (Khalil et al., 1999; Montzka and Fraser, 2003). Microbial CH3Cl degradation in soils may be a relevant additional global sink (McAnulla et al., 2001; Harper et al., 2003; Miller et al., 2004; Jaeger et al., 2018a), but its impact on the global CH3Cl budget is still highly uncertain. The microbial CH3Cl soil sink strength has been estimated to range from 0.1 to 1.6 Tg yr−1 (Harper et al., 2003; Keppler et al., 2005; Carpenter et al., 2014). Moreover, small proportions of tropospheric CH3Cl are lost to the stratosphere (146 Gg yr−1, 1 Gg = 109 g) and to cold polar oceans (370 Gg yr−1) though oceans in total are a net source (Carpenter et al., 2014). Loss of tropospheric CH3Cl to the stratosphere is a result of turbulent mixing and the transport process itself is not thought to cause substantial isotope fractionation (Thompson et al., 2002).
A potentially powerful tool in the investigation of the budgets of atmospheric volatile organic compounds is the use of stable isotope ratios (Brenninkmeijer et al., 2003; Gensch et al., 2014). The general approach is that the atmospheric isotope ratio of a compound (e.g., CH3Cl) is considered to be equal the sum of isotopic fluxes from all sources corrected for kinetic isotopic fractionations that happen in sink processes:
where δ2Hatm and δ2H are the hydrogen isotope values of CH3Cl in the atmosphere and of the different sources i in per mil. Φi and Φj are the CH3Cl flux fraction for each source and sink. εj is the isotope fractionation of each sink j in per mil.
The isotopic composition of atmospheric compounds might be altered by the kinetic isotope effects of physical, chemical or biological loss processes. The kinetic isotope effect (KIE) is usually defined as
where k1 and k2 are the reaction rate constants for loss of the lighter and the heavier isotopologues, respectively. The KIE is typically expressed as isotope fractionation ε (also termed isotope enrichment constant) or isotope fractionation constant α.
First approaches of an isotope mass balance regarding stable carbon isotopes of CH3Cl have been provided by Keppler et al. (2005) and Saito and Yokouchi (2008). Several studies have investigated the stable carbon isotope source signature of CH3Cl produced via biotic and abiotic processes; however, for a more detailed overview we refer readers to the studies of Keppler et al. (2005) and Saito and Yokouchi (2008). Moreover, researchers have measured the KIE of stable carbon isotopes of CH3Cl during oxidation and during biodegradation by bacterial isolates (Miller et al., 2001; Nadalig et al., 2013; Nadalig et al., 2014), and in soils under laboratory conditions (Miller et al., 2004; Jaeger et al., 2018a). The first, and so far, only available analysis of the KIE for reaction of CH3Cl with OH has been reported by Gola et al. (2005) and this revealed unexpectedly large stable carbon isotope fractionation. The experiments were carried out in a smog chamber using long path Fourier transform infrared spectroscopy (FTIR) detection. However, we consider it important to confirm this result using another measurement technique such as stable isotope ratio mass spectrometry (IRMS).
So far most isotopic investigations of CH3Cl have focused on stable carbon isotope measurements, but stable hydrogen isotope measurements including both sources and sinks of CH3Cl have also recently become available (Greule et al., 2012; Nadalig et al., 2013, 2014; Jaeger et al., 2018a, b). Moreover, relative rate experiments have been carried out for three isotopologues of CH3Cl and their reactions with Cl and OH. The OH and Cl reaction rates of CH2DCl were measured by long-path FTIR spectroscopy relative to CH3Cl at 298 ± 2 K and 1 atm (Sellevåg et al., 2006; Table 1).
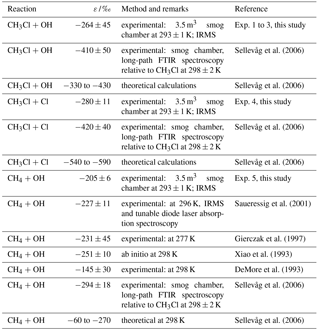
Table 1Reported hydrogen isotope enrichment constants for the reaction of CH3Cl with OH radicals and with Cl atoms and the reaction of CH4 with OH radicals.
In this manuscript, using a 3.5 m3 Teflon smog chamber and IRMS measurements, we present results from kinetic studies of the hydrogen isotope fractionation in the atmospheric OH and Cl loss processes of CH3Cl. Furthermore, we also measured the isotope fractionation for the reaction between methane (CH4) and OH using a similar experimental design and compared this value with those from previous studies.
2.1 Smog chamber experiments with chloromethane
The isotope fractionation experiments were performed in a 3.5 m3 Teflon smog chamber (fluorinated ethylene propylene, FEP 200A, DuPont, Wilmington, DE, USA) with initial CH3Cl mixing ratio of 5 to 10 parts per million by volume (ppmv). Atomic chlorine was generated via photolysis of molecular chlorine (Cl2; Rießner Gase, 0.971 % Cl2 in N2) by a solar simulator with an actinic flux comparable to the sun in mid-summer in Germany (Bleicher et al., 2014). Hydroxyl radicals were generated via the photolysis of ozone (O3) at 253.7 nm in the presence of water vapor (RH = 70 %; produced by double-distilled water in a three-neck bottle humidifier) and/or H2. To obtain efficient OH formation, a Philips TUV lamp T8 (55 W) was coated with Teflon film (FEP 200) and mounted inside the smog chamber. O3 was monitored by a chemiluminescence analyzer (UPK 8001). The chamber was continuously flushed with purified, hydrocarbon-free zero air (zero air generator, cmc Instruments, < 1 ppbv of O3, < 500 pptv NOx, < 100 ppbv of CH4) at a rate of 4 L min−1 to maintain a slight overpressure of 0.5–1 Pa logged with a differential pressure sensor (Kalinsky Elektronik DS1). The quality of the air inside the chamber in terms of possible contamination was controlled by monitoring NO and NOx (EcoPhysics CLD 88p, coupled with a photolytic converter, EcoPhysics PLC 860). Perfluorohexane (PFH) with an initial mixing ratio of ∼ 25 parts per billion by volume (ppbv) was used as an internal standard to correct the resulting concentrations for dilution. The temperature was set to 20 ± 1 ∘C and monitored, together with the relative humidity, by a Teflon-cased sensor (Rotronic, HC2-IC102). To guarantee constant mixing and small temperature gradients, a Teflon fan was mounted and operated inside the chamber. More detailed specification of the smog chamber can be found elsewhere (e.g., Wittmer et al., 2015). The mixing ratios of CH3Cl and PFH were quantified by a Hewlett Packard HP 6890 gas chromatograph coupled to a MSD 5973 mass spectrometer (GC-MS, Agilent Technologies, Palo Alto, CA) with a time resolution of 15 min throughout the experiments. Two CH3Cl reference gases from Linde (1006 ± 12 ppmv diluted in N2) and Scott (1 ppmv) were used for calibration. The abundance of CH3Cl relative to PFH was used to calculate the remaining fraction of CH3Cl (Eq. 4). The relative standard deviation of this procedure was determined prior to each experiment and also during the control experiment and ranged between 1.3 and 1.9 %. Aliquots (5 mL) were withdrawn from the chamber with a gas-tight syringe, injected into a stream of He (30 mL min−1) and directed to a pre-concentration unit that was attached to the GC-MS. The pre-concentration unit consisted of a simple eight-port valve (VICI Valco) equipped with two cryotraps made of fused silica, which were immersed in liquid nitrogen for trapping the analytes. Prior to each sample measurement, a gaseous standard (5 mL of 100 ppmv CH3Cl in N2) was measured. Figure 1 shows the design of the smog chamber used in our experiments.
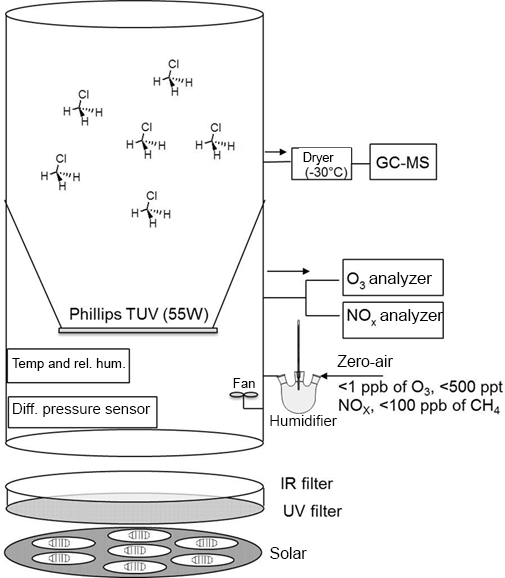
In the CH3Cl and OH experiments (1 to 3) 2000 ppmv H2 was used to scavenge chlorine atoms originating from the photolysis or oxidation of formyl chloride (HCOCl), which forms as an intermediate in the reaction cascade. Under the experimental conditions typically more than 70 % of the CH3Cl was degraded within 6 to 10 h. From each experiment (CH3Cl + OH and CH3Cl + Cl) 10 to 15 canister samples (2 L stainless steel, evacuated < 10−4 mbar) were collected at regular time intervals for subsequent stable hydrogen isotope measurements at Heidelberg University. An overview of the experimental details (Table S1 in the Supplement) and control measurements is provided in the Supplement.
2.2 Smog chamber degradation experiments with methane
The CH4 degradation experiments were carried out under the same conditions as the CH3Cl degradation experiments but without PFH as an internal standard. Instead we used the flushing flow rate of zero air to account for the dilution during the experiment. The initial CH4 mixing ratio was 6 ppmv. Throughout these experiments CH4 and CO2 mixing ratios were monitored with a Picarro G225i cavity ring-down spectrometer directly connected to the chamber. For more details see information provided in the Supplement.
2.3 Stable hydrogen isotope analysis using isotope ratio mass spectrometry
2.3.1 Chloromethane
Stable hydrogen isotope ratios of CH3Cl were measured by an in-house cryogenic pre-concentration unit coupled to a Hewlett Packard HP 6890 gas chromatograph (Agilent Technologies, Palo Alto, CA) and an isotope ratio mass spectrometer (IRMS; Isoprime, Manchester, UK) as described in detail by Greule et al. (2012). Diverging from the method of Greule et al. (2012) a ceramic tube reactor without chromium pellets at 1450 ∘C was instead used for high-temperature conversion (HTC). A tank of high-purity H2 (Alphagaz 2, hydrogen 6.0, Air Liquide, Düsseldorf, Germany) with a δ2H value of ‰ versus the Vienna Standard Mean Ocean Water (VSMOV) was used as the working gas. The conventional delta notation, expressing the isotopic composition of the sample relative to that of VSMOW in per mil is used. All sample δ2H values were measured relative to an in-house working standard of known δ2H value. The CH3Cl working standard was calibrated against IAEA standards VSMOW and SLAP using TC/EA-IRMS (elemental analyzer – isotopic ratio mass spectrometer, IsoLab, Max Planck Institute for Biogeochemistry, Jena, Germany) resulting in a δ2H value of ‰ vs. VSMOW (n=10, 1σ). The H factor, determined daily during this investigation (two different measurement periods), was in the range of 5.75–6.16 (first period) and 8.90–9.21 (second period). The mean precision based on replicate measurements (n=6) of the CH3Cl working standard was 2.1 and 3.8 ‰ for the first and second measurement periods, respectively. Samples were analyzed 3 times (n=3), and the standard deviations (SD) of the measurements were in the range of 1.2 to 103.8 ‰. Lowest SD were observed for samples with lowest δ2H values ( ‰) and highest mixing ratios and higher SD for samples with highest δ2H values ( ‰) and lowest mixing ratios.
Please note that the above-described one-point calibration of the δ2H data might be affected by an additional error (“scale compression”) and particularly might affect the uncertainties of the very positive δ2H values. Unfortunately CH3Cl working standards with distinct isotopic signatures spanning the full range of measured δ2H values (−150 to ‰) are not currently available to eliminate or minimize such an error.
2.3.2 Methane
Stable hydrogen isotope ratios of CH4 were analyzed using an in-house cryogenic pre-concentration unit coupled to a Hewlett Packard HP 6890 gas chromatograph (Agilent Technologies, Palo Alto, CA) and an isotope ratio mass spectrometer (DeltaPlus XL, ThermoQuest Finnigan, Bremen, Germany). The working gas was the same as that used for δ2H analysis of CH3Cl (cf Sect. 2.3.1).
All δ2H values obtained from analysis of CH4 were corrected using two CH4 working standards (isometric instruments, Victoria, Canada) calibrated against IAEA and NIST reference substances (not specified by the company). The calibrated δ2H values of the working standard in ‰ vs. V-SMOW were and ‰.
The H factor determined daily during the 2-week measurement period was in the range 2.38–2.43. The daily average precision based on replicate measurements of the CH4 working standard was 4.9 ‰ (n=7). Samples were analyzed 3 times (n=3), and the SD of the measurements were in the range of 1.4 to 40.9 ‰. Lowest SD were observed for samples with lowest δ2H values ( ‰) and highest mixing ratios and higher SD for samples with highest δ2H values ( ‰) and lowest mixing ratios.
2.4 Kinetic isotope effect, fractionation constant α and the isotope enrichment constant ε
In this study the isotope fractionation constant α and the isotope enrichment constant ε are derived from the slope of the Rayleigh plot according to Clark and Fritz (1997) and Elsner et al. (2005) and Eq. (2):
where Rt and R0 are the 2H / 1H ratios in CH3Cl or CH4 at the different time points and time zero, respectively, and f is the remaining CH3Cl or CH4 fraction at the different time points. Negative values of ε indicates that the remaining CH3Cl or CH4 is enriched in the heavier isotope and corresponds to a α < 1, meaning that over the entire experiment, the heavier CH2DCl or CH3D reacts by this factor more slowly than the lighter CH3Cl or CH4.
The kinetic isotope effect is then calculated as
To correct for ongoing analyte dilution the remaining fraction f has been calculated as follows
where cx0 and cxt are the mixing ratios of CH3Cl at time zero and time t and ci0 and cit are the respective concentrations of the internal standard PFH.
Three experiments of CH3Cl degradation with OH were performed between 25 February 2014 and 3 February 2015. Under the experimental conditions (see Sect. 2 and Supplement) more than 70 % of the CH3Cl was degraded within 6 to 10 h. The results from these experiments are shown in Fig. 2. Both the trends of changes in δ2H values of CH3Cl as well as the remaining fraction of CH3Cl observed in the three independent experiments are in good agreement (Fig. 2a). The calculated ε values for experiments 1 to 3 are , and ‰, respectively (Fig. 2b), with a correlation coefficient R2 of the slope of the regression line of 0.99 for all three experiments.
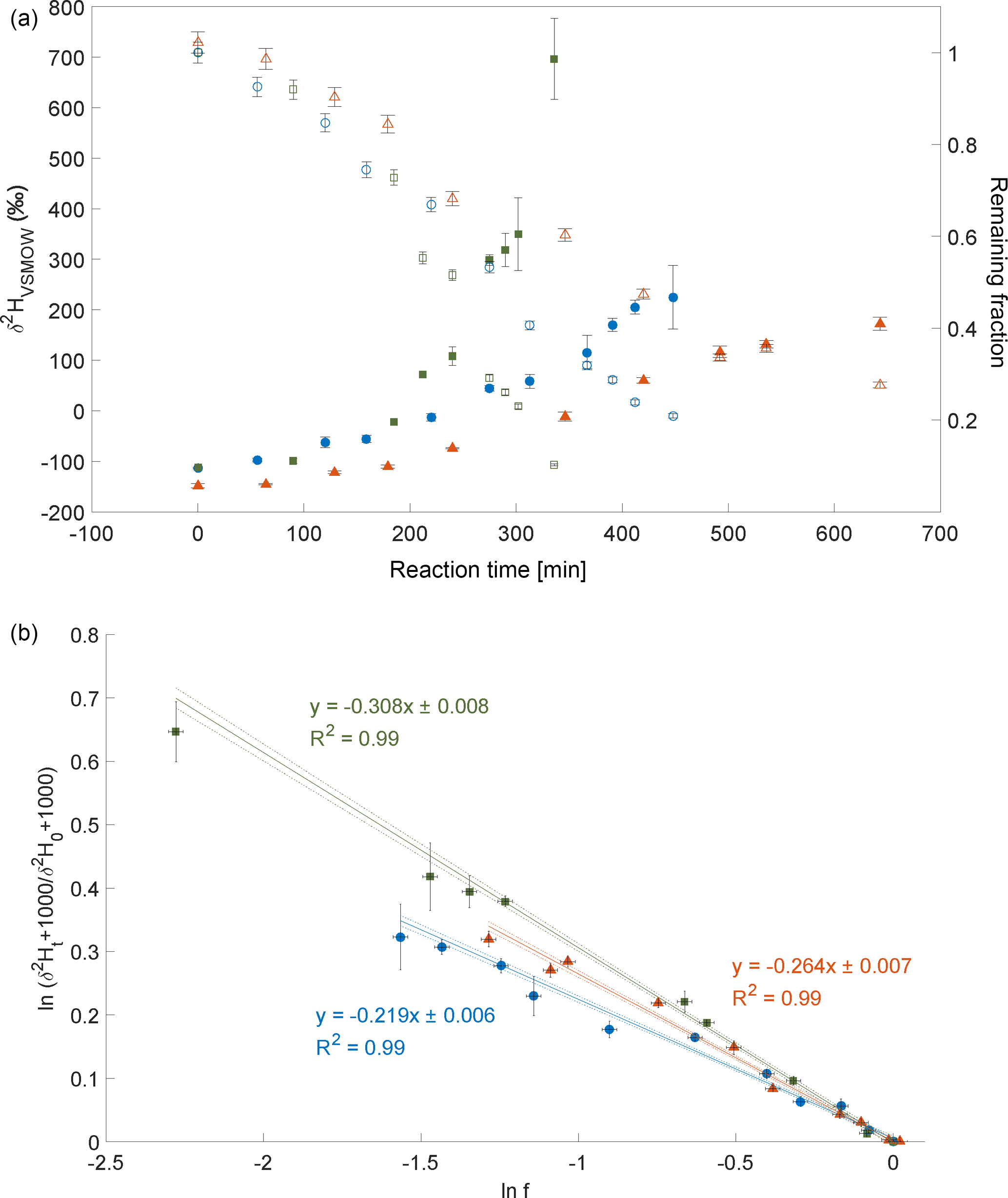
Figure 2Reaction of CH3Cl and OH. Three independent experiments (triangles, dots and squares) were carried out using an initial mixing ratio of 5 to 10 ppmv CH3Cl. More than 70 % of the CH3Cl was degraded within 6 to 10 h. (a) Measured δ2H values (filled circles, triangles and squares) of CH3Cl versus residual fraction (open circles, triangles and squares) of CH3Cl (calculated from changes of CH3Cl and PFH). Error bars of δ2H value of CH3Cl indicate the standard deviation (SD) of the mean of three replicate measurements. Some error bars lie within the symbol. (b) Rayleigh plot (Eq. 3). Error bars were calculated by error propagation including uncertainties in δ2H values of CH3Cl and the remaining fraction. Dashed lines represent 95 % confidence intervals of the linear regressions (bold lines).
The CH3Cl degradation with Cl experiment was conducted on 18 February 2014. Here, over 90 % of CH3Cl was degraded during reaction with Cl radicals within 7 to 8 h (Fig. 3a). The calculated ε of experiment 3 is ‰ (Fig. 3b) with a correlation coefficient of the slope of the regression line of 0.99. Due to limited analytical resources it was not possible to repeat this experiment.
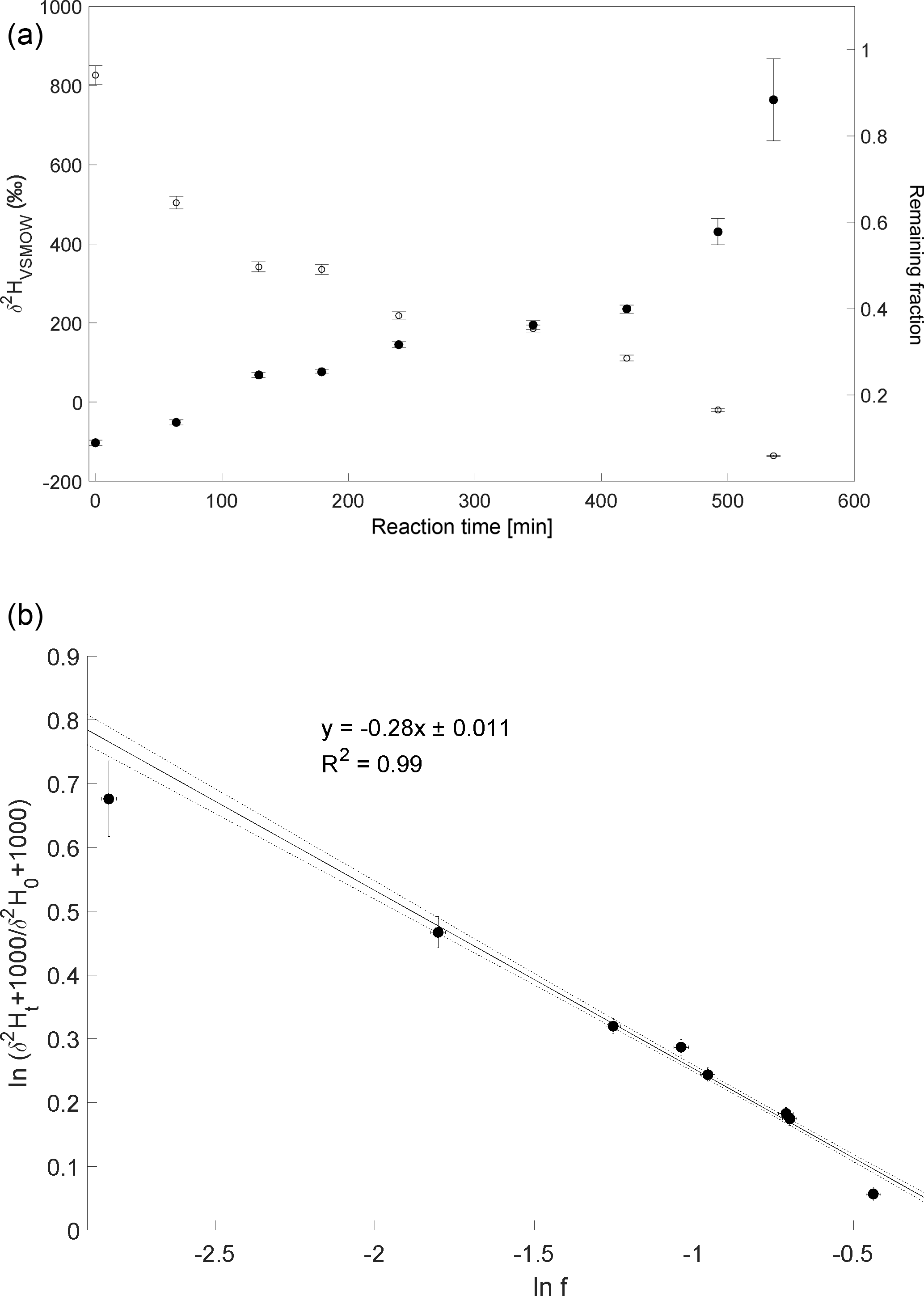
Figure 3Reaction of CH3Cl and Cl. Initial mixing ratio of CH3Cl was ∼ 10 ppmv. More than 90 % of the CH3Cl was degraded within 7 to 8 h. (a) Measured δ2H values (filled circles) of CH3Cl versus residual fraction (open diamonds) CH3Cl. Error bars of δ2H values of CH3Cl indicate the standard deviation (SD) of the mean of three replicate measurements. Some error bars lie within the symbol. (b) Rayleigh plot (Eq. 3). Data are expressed as the mean ± standard error of the mean, n=3. Error bars were calculated by error propagation including uncertainties in δ2H values of CH3Cl. Dashed lines represent 95 % confidence intervals of the linear regressions (bold line).
The experiment to determine the isotope enrichment constant of the degradation of CH4 by hydroxyl radicals was conducted on 2 February 2015. Over 80 % of CH4 was degraded during reaction with OH radicals within 7 h (Fig. 4a). The calculated ε of experiment 4 is ‰ (Fig. 4b) with a correlation coefficient of the slope of the regression line of 0.99.
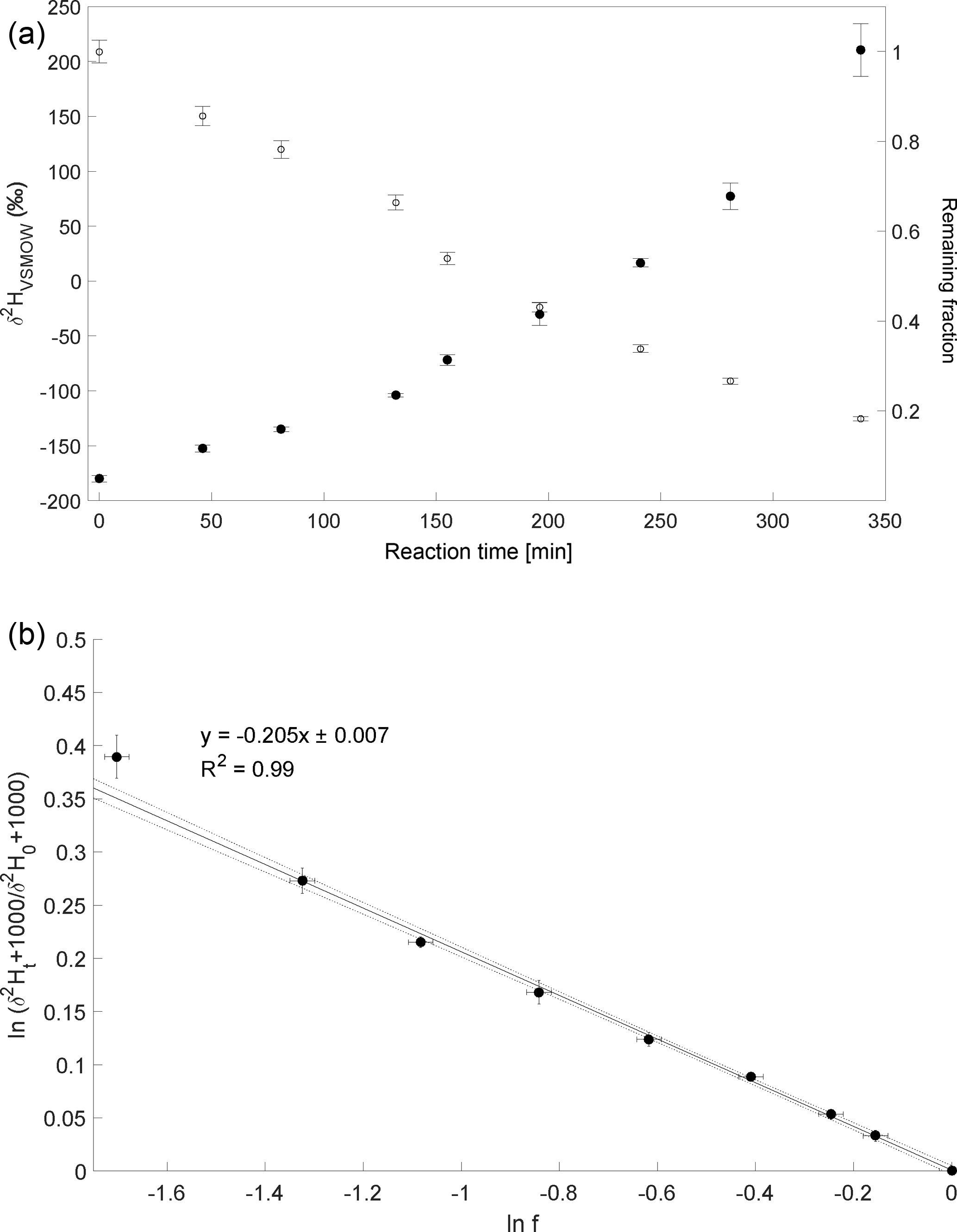
Figure 4Reaction of CH4 and OH. Initial mixing ratio of CH4 was ∼ 6 ppmv. More than 80 % of the CH4 was degraded within 7 h. (a) Measured δ2H values of CH4 versus residual fraction of CH4. Error bars of δ2H values of CH4 indicate the standard deviation (SD) of the mean of three replicate measurements. Some error bars lie within the symbol. (b) Rayleigh plot (Eq. 3). Error bars were calculated by error propagation including uncertainties in δ2H values of CH4 and the remaining fraction. Dashed lines represent 95 % confidence intervals of the linear regressions (bold line).
Chloromethane reacts with both hydroxyl and chlorine radicals in the atmosphere. The first degradation step of CH3Cl in both reactions is the abstraction of a hydrogen atom to yield CH2Cl and H2O or HCl, respectively (Spence et al., 1976; Khalil and Rasmussen, 1999). In both reactions hydrogen is directly present in the reacting bond, and thus influenced by the so-called primary isotope effect (Elsner et al., 2005). Particularly for hydrogen these primary kinetic isotope effects are in general large as they involve a large change in relative mass of the atoms being abstracted. In the following we would like to discuss and compare our results with (i) previous work conducted by Sellevåg et al. (2006), (ii) with OH degradation experiments of CH4 and (iii) with the very recent report of biochemical degradation of CH3Cl in soils and plants (Jaeger et al., 2018a, b).
Although our experimental results show relatively large hydrogen isotope fractionations with ε values of (mean result from three independent experiments ±SD) and ‰ (mean result from three replicate analytical measurements of the same sample ±SD) for reaction of CH3Cl with OH and Cl radicals, respectively, they are smaller than the isotope fractionations previously measured and theoretically calculated by Sellevåg et al. (2006; Table 1). These researchers employed smog chamber experiments at 298 K and used FTIR measurements to determine the stable hydrogen isotope fractionation of CH3Cl and reported ε values of −410 and −420 ‰ for the reaction of CH3Cl with OH and Cl radicals, respectively. They also performed theoretical calculations of ε for the reactions of CH2DCl with OH and Cl radicals and reported ε values in the range of −330 to −430 and −540 to −590 ‰ , respectively (Table 1). Whilst we do not know the reasons for the discrepancies in the experimental ε values observed here and those reported by Sellevåg et al. (2006), we suggest that they may be due to different measurement techniques employed in each of the studies. For further discussion regarding differences of the experimental and analytical design and protocols of the two studies we refer the reader to the Supplement. However, we also conducted similar smog chamber experiments for the degradation of CH4 with hydroxyl radicals (see Sect. 2 and Fig. 4) and calculated an ε value of ‰ for the reaction of CH4 with OH radicals at a temperature of 293 ± 1 K. In Table 1 we compare our results with those from a number of previous studies (Saueressig et al., 2001; Sellevåg et al., 2006; DeMore, 1993; Gierczak et al., 1997; Xiao et al., 1993), which were conducted at temperatures ranging from 277 to 298 K (Table 1). The ε values for the reaction of CH4 with OH radicals from all studies ranged from −145 to −294 ‰ with a mean value of ‰ and the most negative ε value of ‰ reported by Sellevåg et al. (2006). The ε value found in this study ( ‰) was in good agreement with previous experimentally reported values conducted at similar temperatures. This finding gave us confidence that our experimental design and the measurements made using GC-IRMS were reliable.
Compared to primary isotope effects, changes in bonding are much smaller in the case of secondary isotope effects, where positions adjacent to the reacting bond are only slightly affected by the proximity to the reaction center (Elsner et al., 2005; Kirsch, 1977). It was suggested that for the same element, secondary isotope effects are generally at least 1 order of magnitude smaller than primary isotope effects (Kirsch, 1977; Westaway, 1987; Merrigan et al., 1999).
We therefore compared our results from chemical degradation experiments with those from recently reported biochemical degradation experiments (Jaeger et al., 2018a, b). So far, the only known pathway for biochemical consumption of CH3Cl is corrinoid- and tetrahydrofolate-dependent and is termed cmu (abbreviation for chloro methane utilization). This pathway was characterized in detail for the aerobic facultative methylotrophic strain Methylobacterium extorquens CM4 (Vannelli et al., 1999) and involves genes that were also detected in several other chloromethane-degrading strains (Schafer et al., 2007; Nadalig et al., 2011, 2013). During degradation of CH3Cl the methyl group is transferred to a corrinoid cofactor by the protein CmuA. In this case the carbon–chlorine bond of CH3Cl is broken and thus since the hydrogen atoms are adjacent to the reacting bond only a secondary isotope effect would be expected. Indeed, the first ε values reported (Jaeger et al., 2018a, b) for CH3Cl biodegradation by different soils and plants (ferns) are in the range of and ‰, respectively, and thus show considerably smaller kinetic isotope effects than for chemical degradation of CH3Cl by OH and Cl radicals measured in either this study or reported by Sellevåg et al. (2006).
We have performed experiments to measure the hydrogen isotope fractionation of the remaining unreacted CH3Cl following its degradation by hydroxyl and chlorine radicals in a 3.5 m3 Teflon smog chamber at 293 ± 1 K. δ2H values of CH3Cl were measured using GC-IRMS. The calculated isotope fractionations of CH3Cl for the reactions with hydroxyl and with chlorine radicals were found to be smaller than either the experimentally measured (by FTIR) or theoretical values reported by Sellevåg et al. (2006). We also performed degradation experiments of CH4 using the same smog chamber facilities yielding an isotope enrichment constant for the reaction of CH4 with hydroxyl radicals of ‰ which is in good agreement with previously reported results. Although stable hydrogen isotope measurements of CH3Cl sources are still scarce, some recent studies have reported first data on δ2H values of CH3Cl sources and ε values on sinks (Greule et al., 2012; Jaeger et al., 2018, 2018b; Nadalig et al., 2013, 2014).
We have summarized all available information regarding δ2H values of environmental CH3Cl sources and their estimated fluxes in Table 2. Furthermore, the strengths of known CH3Cl sinks and their associated isotope enrichment constants are presented in Table 3. Eventually Fig. 5 displays the global CH3Cl budget showing the known hydrogen isotope signatures of sources and isotope enrichment constants associated with sinks.
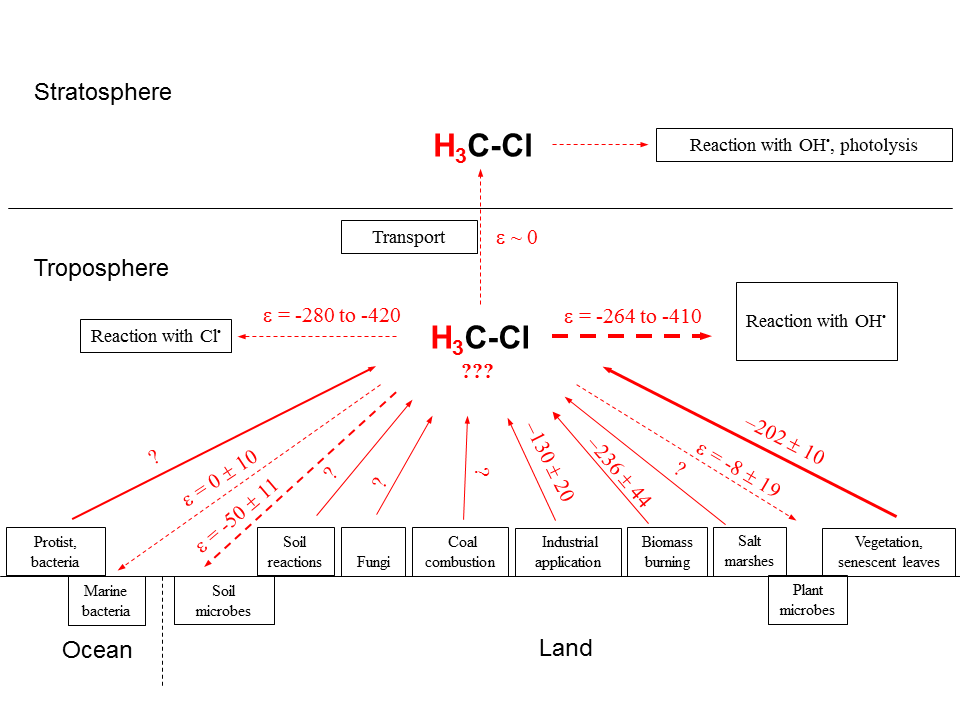
Figure 5Schematic of major sources and sinks involved in the global CH3Cl cycle (modified after Keppler et al., 2005) with known (experimentally determined) corresponding δ2H values and isotope enrichment constants, respectively. Red straight and dashed lines of arrows indicate sources and sinks of CH3Cl, respectively. Size/thickness of arrows indicates strength of fluxes in the environment. Question marks indicate where currently no data exist. All values are given in per mil.
Table 2Known sources and strengths of tropospheric CH3Cl and corresponding δ2H values.
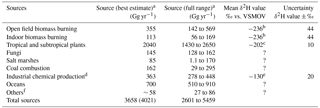
a Values for source (best estimate) and source (full range) were taken from Carpenter and Reimann (2014), except for emissions associated with chemical production by the industry which are from Li et al. (2017). The value shown for total sources in parentheses includes chemical production by the industry. b Greule et al. 2012; please note that all values provided for CH3Cl released from dried plants at elevated temperatures have been corrected by −23 ‰ due to recalibration of the reference gas. c Jaeger et al. (2018b). d Li et al. (2017). e taken from Greule et al. (2012), Nadalig et al. (2013) and Jaeger et al. (2018a, 2018b); please note that values provided by Greule et al. (2012) and Nadalig et al. (2013) for CH3Cl from sources of the chemical industry have been corrected by −23 ‰ due to recalibration of the reference gas. f including mangroves, wetlands, rice paddies and shrublands. ? denotes that no value has been provided.
Table 3Known sinks of tropospheric CH3Cl and the mean isotope enrichment constant ε reported for each.
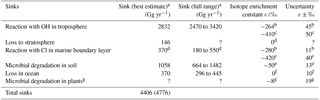
a Values for sink strength (best estimate and full range) were taken from Carpenter and Reimann (2014), except for the value of the reaction with Cl radicals in marine boundary layer and for total sinks shown in parentheses which includes the potential sink strength by Cl radicals in marine boundary layer (Montzka and Fraser, 2003). b this study. c Sellevåg et al. (2006). d Thompson et al. (2002) and discussion in this manuscript. e Jaeger et al. (2018a). f Nadalig et al. (2014). g Jaeger et al. (2018b). ? denotes that no value has been provided.
Our results suggest that stable hydrogen isotope measurements of both sources and sinks of CH3Cl and particularly the observed large kinetic isotope effect of the atmospheric CH3Cl sinks might strongly assist with the refinement of current models of the global atmospheric CH3Cl budget. In contrast to the large hydrogen fractionation of CH3Cl by chemical degradation of OH and Cl radicals, the isotope fractionation of CH3Cl biodegradation is in the range of an order of magnitude lower. This therefore has the possibility of improving our understanding of the global CH3Cl budget once the δ2H value of atmospheric CH3Cl has been measured. The stable hydrogen isotopic composition of tropospheric CH3Cl depends on the isotopic source signatures and the kinetic isotope effects of the sinks, primarily the reaction with OH and consumption by soils and potentially plants.
Several attempts at modeling the global CH3Cl budget using stable carbon isotope ratios have already been made (Harper et al., 2001, 2003; Thompson et al., 2002; Keppler et al., 2005; Saito and Yokouchi, 2008) but there are still major uncertainties regarding source and sink strengths as well as the respective stable isotope signatures. Therefore, we now suggest combining our knowledge of stable carbon and hydrogen isotopes of CH3Cl in the environment. Such a two-dimensional (2-D) stable isotope approach of hydrogen and carbon can be used to better understand the processes of CH3Cl biodegradation and formation. Furthermore, when this approach is combined with CH3Cl flux estimates it could help to better constrain the strength of CH3Cl sinks and sources within the global CH3Cl budget (Nadalig et al., 2014; Jaeger et al., 2018b).
We note that currently no data are available for the δ2H value of atmospheric CH3Cl. Although it will be a massive analytical challenge to obtain this value, we strongly suspect that it would likely lead to a better-refined isotopic mass balance for atmospheric CH3Cl and thus to a better understanding of the global CH3Cl budget.
The data used in this publication are available to the community and can be accessed by request to the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-6625-2018-supplement.
The authors declare that they have no conflict of interest.
This study was supported by DFG (KE 884/8-1; KE 884/8-2, KE 884/10-1) and by
the DFG research unit 763 “Natural Halogenation Processes in the Environment
– Atmosphere and Soil” (KE 884/7-1, SCHO 286/7-2, ZE 792/5-2). We further
acknowledge the German Federal Ministry of Education and Research (BMBF) for
funding within SOPRAN “Surface Ocean Processes in the Anthropocene” (grants
03F0611E and 03F0662E). We thank John Hamilton and Carl Brenninkmeijer for
comments on an earlier version of the manuscript and Daniela Polag for
statistical evaluation of the data.
The article processing charges for this open-access
publication were covered by the Max Planck Society.
Edited by:
Sergey A. Nizkorodov
Reviewed by: Matthew Johnson and two anonymous referees
Bleicher, S., Buxmann, J. C., Sander, R., Riedel, T. P., Thornton, J. A., Platt, U., and Zetzsch, C.: The influence of nitrogen oxides on the activation of bromide and chloride in salt aerosol, Atmos. Chem. Phys. Discuss., 14, 10135–10166, https://doi.org/10.5194/acpd-14-10135-2014, 2014.
Brenninkmeijer, C. A. M., Janssen, C., Kaiser, J., Röckmann, T., Rhee, T. S., and Assonov, S. S.: Isotope Effects in the Chemistry of Atmospheric Trace Compounds, Chem. Rev., 103, 5125–5162, https://doi.org/10.1021/cr020644k, 2003.
Carpenter, L. J., Reimann, S., Burkholder, J. B., Clerbaux, C., Hall, B., Hossaini, R., Laube, J., and Yvon-Lewis, S.: Chapter 1: Update on Ozone-Depleting Substances (ODSs) and Other Gases of Interest to the Montreal Protocol, in: Scientific Assessment of Ozone Depletion, Global Ozone Research and Monitoring Project Report, World Meteorological Organization (WMO), 21–125, 2014.
Clark, I. and Fritz, P.: Environmental isotopes in hydrogeology, Lewis Publishers, New York, 328 pp., 1997.
DeMore, W. B.: Rate constant ratio for the reaction of OH with CH3D and CH4, J. Phys. Chem., 97, 8564–8566, 1993.
Derendorp, L., Holzinger, R., Wishkerman, A., Keppler, F., and Rockmann, T.: Methyl chloride and C(2)-C(5) hydrocarbon emissions from dry leaf litter and their dependence on temperature, Atmos. Environ., 45, 3112–3119, https://doi.org/10.1016/j.atmosenv.2011.03.016, 2011.
Elsner, M., Zwank, L., Hunkeler, D., and Schwarzenbach, R. P.: A new concept linking observable stable isotope fractionation to transformation pathways of organic pollutants, Environ. Sci. Technol., 39, 6896–6916, 2005.
Gensch, I., Kiendler-Scharr, A., and Rudolph, J.: Isotope ratio studies of atmospheric organic compounds: Principles, methods, applications and potential, Int. J. Mass Spectrom., 365–366, 206–221, https://doi.org/10.1016/j.ijms.2014.02.004, 2014.
Gierczak, T., Talukdar, R. K., Herndon, S. C., Vaghjiani, G. L., and Ravishankara, A. R.: Rate Coefficients for the Reactions of Hydroxyl Radicals with Methane and Deuterated Methanes, J. Phys. Chem. A, 101, 3125–3134, https://doi.org/10.1021/jp963892r, 1997.
Gola, A. A., D'Anna, B., Feilberg, K. L., Sellevåg, S. R., Bache-Andreassen, L., and Nielsen, C. J.: Kinetic isotope effects in the gas phase reactions of OH and Cl with CH3Cl, CD3Cl, and 13CH3Cl, Atmos. Chem. Phys., 5, 2395–2402, https://doi.org/10.5194/acp-5-2395-2005, 2005.
Greule, M., Huber, S. G., and Keppler, F.: Stable hydrogen-isotope analysis of methyl chloride emitted from heated halophytic plants, Atmos. Environ., 62, 584–592, https://doi.org/10.1016/j.atmosenv.2012.09.007, 2012.
Hamilton, J. T. G., McRoberts, W. C., Keppler, F., Kalin, R. M., and Harper, D. B.: Chloride methylation by plant pectin: An efficient environmentally significant process, Science, 301, 206–209, 2003.
Harper, D. B.: Halomethane from halide ion – a highly efficient fungal conversion of environemnetal significance, Nature, 315, 55–57, 1985.
Harper, D. B., Kalin, R. M., Hamilton, J. T. G., and Lamb, C.: Carbon isotope ratios for chloromethane of biological origin: Potential tool in determining biological emissions, Environ. Sci. Technol., 35, 3616–3619, 2001.
Harper, D. B., Hamilton, J. T. G., Ducrocq, V., Kennedy, J. T., Downey, A., and Kalin, R. M.: The distinctive isotopic signature of plant-derived chloromethane: possible application in constraining the atmospheric chloromethane budget, Chemosphere, 52, 433–436, 2003.
Jaeger, N., Besaury, l., Kröber, E., Delort, A.-M., Greule, M., Lenhart, K., Nadalig, T., Vuilleumier, S., Amato, P., Kolb, S., Bringel, F., and Keppler, F.: Chloromethane degradation in soils – a combined microbial and two-dimensional stable isotope approach, J. Environ. Qual., 47, 254–262, 2018a.
Jaeger, N., Besaury, L., Röhling, A. N., Koch, F., Delort, A. M., Gasc, C., Greule, M., Kolb, S., Nadalig, T., Peyret, P., Vuilleumier, S., Amato, P., Bringel, F., and Keppler, F.: Chloromethane formation and degradation in the fern phyllosphere, Sci. Total Environ., 634, 1278–1287, https://doi.org/10.1016/j.scitotenv.2018.03.316, 2018b.
Keppler, F., Eiden, R., Niedan, V., Pracht, J., and Scholer, H. F.: Halocarbons produced by natural oxidation processes during degradation of organic matter, Nature, 403, 298–301, 2000.
Keppler, F., Harper, D. B., Röckmann, T., Moore, R. M., and Hamilton, J. T. G.: New insight into the atmospheric chloromethane budget gained using stable carbon isotope ratios, Atmos. Chem. Phys., 5, 2403–2411, https://doi.org/10.5194/acp-5-2403-2005, 2005.
Keppler, F., Fischer, J., Sattler, T., Polag, D., Jaeger, N., Schöler, H. F., and Greule, M.: Chloromethane emissions in human breath, Sci. Total Environ., 605–606, 405–410, https://doi.org/10.1016/j.scitotenv.2017.06.202, 2017.
Khalil, M. A. K. and Rasmussen, R. A.: Atmospheric methyl chloride, Atmos. Environ., 33, 1305–1321, https://doi.org/10.1016/S1352-2310(98)00234-9, 1999.
Khalil, M. A. K., Moore, R. M., Harper, D. B., Lobert, J. M., Erickson, D. J., Koropalov, V., Sturges, W. T., and Keene, W. C.: Natural emissions of chlorine-containing gases: Reactive Chlorine Emissions Inventory, J. Geophys. Res.-Atmos., 104, 8333–8346, 1999.
Kirsch, J. F.: Isotope effects on enzyme-catalyzed reactions, edited by: Cleland, W. W., O'Leary, M. H., and Northrop, D. B., University Park Press, Baltimore, London, Tokyo, 100–121, 1977.
Kolusu, S. R., Schlünzen, K. H., Grawe, D., and Seifert, R.: Chloromethane and dichloromethane in the tropical Atlantic Ocean, Atmos. Environ., 150, 417–424, 2017.
Li, S., Park, M.-K., Jo, C. O., and Park, S.: Emission estimates of methyl chloride from industrial sources in China based on high frequency atmospheric observations, J. Atmos. Chem., 74, 227–243, https://doi.org/10.1007/s10874-016-9354-4, 2017.
McAnulla, C., McDonald, I. R., and Murrell, J. C.: Methyl chloride utilising bacteria are ubiquitous in the natural environment, FEMS Microbiol. Lett., 201, 151–155, https://doi.org/10.1111/j.1574-6968.2001.tb10749.x, 2001.
Merrigan, S. R., Le Gloahec, V. N., Smith, J. A., Barton, D. H. R., and Singleton, D. A.: Separation of the primary and secondary kinetic isotope effects at a reactive center using starting material reactivities. Application to the FeCl3-Catalyzed oxidation of C-H bonds with tert-butyl hydroperoxide, Tetrahedron Lett., 40, 3847–3850, https://doi.org/10.1016/S0040-4039(99)00637-1, 1999.
Miller, L. G., Kalin, R. M., McCauley, S. E., Hamilton, J. T. G., Harper, D. B., Millet, D. B., Oremland, R. S., and Goldstein, A. H.: Large carbon isotope fractionation associated with oxidation of methyl halides by methylotrophic bacteria, P. Natl. Acad. Sci. USA, 98, 5833–5837, 2001.
Miller, L. G., Warner, K. L., Baesman, S. M., Oremland, R. S., McDonald, I. R., Radajewski, S., and Murrell, J. C.: Degradation of methyl bromide and methyl chloride in soil microcosms: Use of stable C isotope fractionation and stable isotope probing to identify reactions and the responsible microorganisms, Geochim. Cosmochim. Acta, 68, 3271–3283, 2004.
Montzka, S. A. and Fraser, P.: Controlled substances and other source gases, Chapter 1 in Scientific Assessment of Ozone Depletion: 2002, World Meteorological Organization, Geneva, 2003.
Moore, R. M., Groszko, W., and Niven, S. J.: Ocean-atmosphere exchange of methyl chloride: Results from NW Atlantic and Pacific Ocean studies, J. Geophys. Res.-Oceans, 101, 28529–28538, https://doi.org/10.1029/96jc02915, 1996.
Nadalig, T., Farhan Ul Haque, M., Roselli, S., Schaller, H., Bringel, F., and Vuilleumier, S.: Detection and isolation of chloromethane-degrading bacteria from the Arabidopsis thaliana phyllosphere, and characterization of chloromethane utilization genes, FEMS Microbiol. Ecol., 77, 438–448, https://doi.org/10.1111/j.1574-6941.2011.01125.x, 2011.
Nadalig, T., Greule, M., Bringel, F., Vuilleumier, S., and Keppler, F.: Hydrogen and carbon isotope fractionation during degradation of chloromethane by methylotrophic bacteria, Microbiologyopen, 2, 893–900, https://doi.org/10.1002/mbo3.124, 2013.
Nadalig, T., Greule, M., Bringel, F., Keppler, F., and Vuilleumier, S.: Probing the diversity of chloromethane-degrading bacteria by comparative genomics and isotopic fractionation, Front. Microbiol., 5, 523, https://doi.org/10.3389/fmicb.2014.00523, 2014.
Redeker, K. R., Wang, N.-Y., Low, J. C., McMillan, A., Tyler, S. C., and Cicerone, R. J.: Emissions of Methyl Halides and Methane from Rice Paddies, Science, 290, 966–969, https://doi.org/10.1126/science.290.5493.966, 2000.
Rhew, R. C., Miller, B. R., and Weiss, R. F.: Natural methyl bromide and methyl chloride emissions from coastal salt marshes, Nature, 403, 292–295, https://doi.org/10.1038/35002043, 2000.
Rhew, R. C., Aydin, M., and Saltzman, E. S.: Measuring terrestrial fluxes of methyl chloride and methyl bromide using a stable isotope tracer technique, Geophys. Res. Lett., 30, 2103, https://doi.org/10.1029/2003GL018160, 2003.
Saito, T. and Yokouchi, Y.: Stable carbon isotope ratio of methyl chloride emitted from glasshouse-grown tropical plants and its implication for the global methyl chloride budget, Geophys. Res. Lett., 35, l08807, https://doi.org/10.1029/2007gl032736, 2008.
Saueressig, G., Crowley, J. N., Bergamaschi, P., Brühl, C., Brenninkmeijer, C. A. M., and Fischer, H.: Carbon 13 and D kinetic isotope effects in the reactions of CH4 with O(1D) and OH: New laboratory measurements and their implications for the isotopic composition of stratospheric methane, J. Geophys. Res.-Atmos., 106, 23127–23138, 10.1029/2000JD000120, 2001.
Schafer, H., Miller, L. G., Oremland, R. S., and Murrell, J. C.: Bacterial cycling of methyl halides, Adv. Appl. Microbiol., 61, 307–346, 2007.
Sellevåg, S. R., Nyman, G., and Nielsen, C. J.: Study of the Carbon-13 and Deuterium Kinetic Isotope Effects in the Cl and OH Reactions of CH4 and CH3Cl, J. Phys. Chem. A, 110, 141–152, https://doi.org/10.1021/jp0549778, 2006.
Spence, J. W., Hanst, P. L., and Gay, B. W.: Atmospheric Oxidation of Methyl Chloride Methylene Chloride, and Chloroform, JAPCA J. Air. Waste Ma., 26, 994–996, https://doi.org/10.1080/00022470.1976.10470354, 1976.
Thompson, A. E., Anderson, R. S., Rudolph, J., and Huang, L.: Stable carbon isotope signatures of background tropospheric chloromethane and CFC113, Biogeochemistry, 60, 191–211, 2002.
Umezawa, T., Baker, A. K., Brenninkmeijer, C. A. M., Zahn, A., Oram, D. E., and van Velthoven, P. F. J.: Methyl chloride as a tracer of tropical tropospheric air in the lowermost stratosphere inferred from IAGOS-CARIBIC passenger aircraft measurements, J. Geophys. Res.-Atmos., 120, 12313–12326, https://doi.org/10.1002/2015JD023729, 2015.
Vannelli, T., Messmer, M., Studer, A., Vuilleumier, S., and Leisinger, T.: A corrinoid-dependent catabolic pathway for growth of a Methylobacterium strain with chloromethane, P. Natl. Acad. Sci. USA, 96, 4615–4620, https://doi.org/10.1073/pnas.96.8.4615, 1999.
Westaway, K. C.: In Isotopes in organic chemistry, edited by: Buncel, E., and Lee, C. C., Elsevier, New York, 275–392, 1987.
Williams, J., Wang, N.-Y., Cicerone, R. J., Yagi, K., Kurihara, M., and Terada, F.: Atmospheric methyl halides and dimethyl sulfide from cattle, Glob. Biogeochem. Cycle, 13, 485–491, https://doi.org/10.1029/1998GB900010, 1999.
Wittmer, J., Bleicher, S., and Zetzsch, C.: Iron(III)-induced activation of chloride and bromide from modeled salt pans, J. Phys. Chem. A, 119, 4373–4385, https://doi.org/10.1021/jp508006s, 2015.
WMO: Scientific Assessment of Ozone Depletion: 2010, Global Ozone Research and Monitoring Project-Report No. 52, 516 pp., Geneva, Switzerland, 2011.
Xiao, Y., Tanaka, N., and Lasaga, A.: An evaluation of hydrogen kinetic isotope effect in the reaction of CH4 with OH free radical (abstract), in: Eos Trans. AGU, 74, Spring Meet. Suppl., 71, 1993.
Yokouchi, Y., Ikeda, M., Inuzuka, Y., and Yukawa, T.: Strong emission of methyl chloride from tropical plants, Nature, 416, 163–165, 2002.
Yokouchi, Y., Saito, T., Ishigaki, C., and Aramoto, M.: Identification of methyl chloride-emitting plants and atmospheric measurements on a subtropical island, Chemosphere, 69, 549–553, 2007.