the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 13 Apr 2018
| 13 Apr 2018
Continuous non-marine inputs of per- and polyfluoroalkyl substances to the High Arctic: a multi-decadal temporal record
Heidi M. Pickard
Alison S. Criscitiello
Christine Spencer
Martin J. Sharp
Derek C. G. Muir
Amila O. De Silva
Perfluoroalkyl acids (PFAAs) are persistent, in some cases, bioaccumulative compounds found ubiquitously within the environment. They can be formed from the atmospheric oxidation of volatile precursor compounds and undergo long-range transport (LRT) through the atmosphere and ocean to remote locations. Ice caps preserve a temporal record of PFAA deposition making them useful in studying the atmospheric trends in LRT of PFAAs in polar or mountainous regions, as well as in understanding major pollutant sources and production changes over time. A 15 m ice core representing 38 years of deposition (1977–2015) was collected from the Devon Ice Cap in Nunavut, providing us with the first multi-decadal temporal ice record in PFAA deposition to the Arctic. Ice core samples were concentrated using solid phase extraction and analyzed by liquid and ion chromatography methods. Both perfluoroalkyl carboxylic acids (PFCAs) and perfluoroalkyl sulfonic acids (PFSAs) were detected in the samples, with fluxes ranging from < LOD to 141 ng m−2 yr−1. Our results demonstrate that the PFCAs and perfluorooctane sulfonate (PFOS) have continuous and increasing deposition on the Devon Ice Cap, despite recent North American and international regulations and phase-outs. We propose that this is the result of on-going manufacture, use and emissions of these compounds, their precursors and other newly unidentified compounds in regions outside of North America. By modelling air mass transport densities, and comparing temporal trends in deposition with production changes of possible sources, we find that Eurasian sources, particularly from Continental Asia, are large contributors to the global pollutants impacting the Devon Ice Cap. Comparison of PFAAs to their precursors and correlations of PFCA pairs showed that deposition of PFAAs is dominated by atmospheric formation from volatile precursor sources. Major ion analysis confirmed that marine aerosol inputs are unimportant to the long-range transport mechanisms of these compounds. Assessments of deposition, homologue profiles, ion tracers, air mass transport models, and production and regulation trends allow us to characterize the PFAA depositional profile on the Devon Ice Cap and further understand the LRT mechanisms of these persistent pollutants.
- Article
(3127 KB) -
Supplement
(874 KB) - BibTeX
- EndNote





Per- and polyfluoroalkyl substances (PFASs) are a diverse group of compounds that have been used in surfactants and polymers for over 60 years (Buck et al., 2011). Perfluoroalkyl acids (PFAAs), a sub-group of PFASs, are persistent contaminants that are ubiquitous in the environment. Perfluoroalkyl carboxylic acids (PFCAs) and perfluoroalkyl sulfonic acids (PFSAs) are two of the most widely known and studied groups of PFAAs (Buck et al., 2011; Stock et al., 2007). PFAAs are prevalent in remote locations, such as the Arctic (Butt et al., 2010), due to their ability to undergo long-range transport through the atmosphere and/or the ocean (Prevedouros et al., 2006). Long-range transport can be a combination of both direct transport and indirect formation. With direct transport, PFAAs are directly transported in their carboxylic (PFCA) or sulfonic (PFSA) acid form to remote locations. This can occur through the atmosphere, in the gaseous or particle phase (including with marine aerosols), or via oceanic water currents (Prevedouros et al., 2006). With indirect formation, PFAAs are produced through chemical transformation of PFAS precursors in the atmosphere (Young and Mabury, 2010). These PFAAs are environmentally persistent and longer chain acids (> 6 carbons) have a tendency to bioaccumulate and biomagnify in food webs (Butt et al., 2010; Houde et al., 2006; Scheringer et al., 2014).
In the atmosphere, volatile and semi-volatile precursors such as fluorotelomer alcohols (FTOHs) and N-alkyl perfluoroalkane sulfonamides/sulfonamidoethanols ((N-Alkyl) FASAs/FASEs) undergo oxidation in the gas phase to form PFAAs (D'eon et al., 2006; Ellis et al., 2004; Young and Mabury, 2010). The atmospheric lifetime and persistence of these precursors is long enough to reach remote locations by wind and air transport, before subsequently oxidizing to the corresponding PFAAs and depositing to remote locations (Busch et al., 2010; Young et al., 2007). Once these PFAAs are formed in the gas phase, they undergo wet or dry deposition to the surface. The specifics of these processes have been considered in several modelling studies in the literature (Armitage et al., 2009a, b; Schenker et al., 2008; Wallington et al., 2006; Yarwood et al., 2007).
The long-range transport mechanisms of these compounds can be elucidated through the collection and analysis of remote samples, such as ice core samples. Ice caps receive their contamination solely from atmospheric deposition due to their high elevation, and preserve a temporal record of that deposition. The Devon Ice Cap, located on Devon Island in Nunavut, Canada, was previously sampled for PFAAs in May of both 2006 and 2008 through collection from the sidewall of a snow pit (MacInnis et al., 2017; Young et al., 2007) This ice cap has a high latitude and elevation (Boon et al., 2010) and is not expected to receive any local or oceanic sources of contamination. These previous studies detected PFAAs in snow profiles that spanned a 10–14 year period in deposition. In this study, a 15 m ice core was collected in 2015, allowing us to examine PFAA deposition over a much longer (38 year) period. Within this paper we discuss the following: (1) PFAA deposition and temporal trends; (2) homologue patterns and volatile precursor mechanisms; (3) transport of PFAAs to the Arctic via ion tracer analysis; and (4) PFAA source regions via transport modelling. This work represents the first multi-decadal analysis of PFAAs in an ice core from the summit region (2175 m above mean sea level, m a.m.s.l.) of a large Arctic ice cap.
2.1 Sample collection and sectioning
A 15.5 m ice core was collected from the Devon Ice Cap, Devon Island, Nunavut (75.2∘ N, 82.7∘ W, 2175 m a.m.s.l.) on 17 May 2015. Extensive measures were taken to avoid PFAS contamination during both sample collection and sectioning (i.e. PFAS-free apparel and equipment, methanol-rinsed sampling tools and equipment). Samples were collected using a stainless steel Kovacs ice drill with a 9 cm diameter (Sect. S1 in the Supplement). The samples were separated into 1 m ice core sections, packaged in polyethylene wrap, shipped frozen to the Canada Centre for Inland Waters (CCIW) in Burlington, Ontario, Canada and stored at −35 ∘C prior to sectioning. Cores were sectioned in a −10 ∘C freezer into discrete samples corresponding to individual years. Sectioning was done using stainless steel tools, cleaned with methanol (MeOH; Omnisolv, 99.9 %, EMD, ThermoFisher) before and after each sample. Ice core sections were placed into pre-cleaned 4 L high-density polypropylene bottles and kept frozen at −35 ∘C prior to extraction and analysis.
2.2 Sample preparation and extraction
Sample extraction has been described previously (MacInnis et al., 2017). Briefly, samples were thawed immediately prior to extraction and aliquoted into 500 mL melted volumes for extraction. Sub-samples for extraction were spiked with 30 µL of a surrogate mixture (Table S1 in the Supplement), which acted as the internal standard (IS) to monitor recovery (Table S2). Samples were shaken, sonicated for 10 min, and held for 30 min at room temperature.
Targeted analytes included: trifluoroacetic acid (TFA), perfluoropropionic acid (PFPrA), perfluorobutanoic acid (PFBA), perfluoropentanoic acid (PFPeA), perfluorohexanoic acid (PFHxA), perfluoroheptanoic acid (PFHpA), perfluorooctanoic acid (PFOA), perfluorononanoic acid (PFNA), perfluorodecanoic acid (PFDA), perfluoroundecanoic acid (PFUnDA), perfluorododecanoic acid (PFDoDA), perfluorotridecanoic acid (PFTrDA), perfluorotetradecanoic acid (PFTeDA), perfluorohexadecanoic acid (PFHxDA), perfluorooctadecanoic acid (PFOcDA), perfluorobutane sulfonic acid (PFBS), perfluorohexane sulfonic acid (PFHxS), perfluoroheptane sulfonic acid (PFHpS), perfluorooctane sulfonic acid (PFOS), perfluorodecane sulfonic acid (PFDS), perfluoroethylcyclohexanesulfonate (PFECHS), and perfluorooctanesulfonamide (FOSA).
Samples were concentrated using an OASIS® weak anion exchange solid phase extraction (SPE) cartridge (6 cm3, 150 mg, 30 µm). Cartridges were conditioned prior to sample loading with 5 mL 0.1 % NH4OH∕MeOH, followed by 5 mL MeOH and 5 mL SPE-cleaned HPLC Grade water (Fisher). Following sample concentration, the cartridges were rinsed with 25 mM ammonium acetate buffer acidified to pH 4 with acetic acid, and centrifuged at 4000 rpm for 2 min to remove any residual water. Samples were eluted into two fractions: the first fraction was eluted with 6 mL of MeOH for FOSA, and the second fraction was eluted with 8 mL of 0.1 % NH4OH∕MeOH for PFAAs. Both fractions were evaporated to dryness under a gentle stream of nitrogen and reconstituted in 0.5 mL 50∕50 methanol – water containing the surrogate mixture (Table S1) to monitor matrix effects (Table S3). Reconstituted samples were sonicated for 5 min, vortexed, and transferred to polypropylene vials for analysis.
2.3 Quality assurance and quality control
Previous results from field blanks (HPLC grade water) transported and exposed to the atmosphere in the Canadian Arctic (Resolute Bay, Nunavut) have indicated that the environmental exposure and shipping do not contribute to background PFAS contamination. We have amassed an annual data set on these field blanks (Table S4a–b). Cartridge blanks were used to validate the integrity of the extraction method, and isotopically labelled standards were used to validate recovery and matrix effects. Three samples were extracted in triplicate and one sample in duplicate to evaluate reproducibility. A composite mixture of the Devon Ice Cap samples was prepared for three types of QA/QC measures in triplicate: one sample was spiked with PFASs before extraction, one sample spiked with PFASs after extraction, and the third sample was spiked with the internal standard and processed akin to the larger sample set. The pre-extraction and post-extraction spiked samples were compared to evaluate recovery and matrix effects. No quantifiable PFAA levels were detected in the routinely analyzed method blanks (SPE cartridge blanks, n= 6). Method recoveries for the PFAAs ranged from 79 to 117 % with the exception of PFOcDA (127–225 %). PFOcDA recoveries indicated enhancement of analyte signal due to matrix effects and incorrect recovery and matrix correction due to using MPFHxDA as the internal standard. Since PFOcDA was below detection limit in all ice core samples, this was not explored any further.
Matrix effects were evaluated by comparing the peak area of instrument performance standard (IP) compounds to peak areas at equivalent concentrations in a solvent standard. Recovery was evaluated by comparing the recovered analyte concentration in the spike and recovery sample to the theoretical spiked concentration. Each sample was corrected for recovery and matrix effects by quantifying the concentrations based on relative response to isotopically labelled standards added before extraction. A 15 level calibration curve was employed ranging from 0.02 to 8.5 ng mL−1, along with analytical blanks. Analytical blanks (MeOH) and cartridge blanks were included in the method analysis. The method detection limit (MDL) was based on three times the standard deviation of the cartridge blanks. Most PFAA analytes were not detected in the method blanks and were therefore below the instrument detection limit (IDL; Table S5). The limit of detection (LOD) and limit of quantitation (LOQ) were quantified based on signal-to-noise (S/N) ratios of 3 and 10, respectively (Table S6).
2.4 Sample analysis
Samples were analyzed by ultra performance liquid chromatography (Waters Acquity UPLC I) with tandem mass spectrometry (Waters Xevo® TQ-S, UPLC-MS/MS) detection operated in electrospray negative ionization mode. Two analyses were conducted. For the first PFAA (PFCAs > C4, PFSAs > C4, FOSA) analysis, samples were separated using a C18 column (Waters Acquity UPLC® BEH, 2.1 × 50 mm, 1.7 µm) with a water – methanol 2 mM ammonium acetate gradient method. For the second PFAA (PFCAs < C8, PFOS) analysis, samples were separated using a Shodex RSPak column (2.0 × 150 mm, 5 µm) with a water – methanol 50 mM ammonium acetate method (Tables S7–S8). Analytes were quantified based on relative response to isotopically labelled internal standards (Wellington Laboratories, Guelph, ON).
2.5 Major ion analysis
Sub samples of the sectioned ice core (15 mL) were analyzed for major anions and cations. Anions were measured by ion chromatography with conductivity detection and cations were quantified using inductively coupled plasma and optical emission detection. A range of cations (Na+, K+, Ca2+, Mg2+, Mn2+, Al3+) and other metals (e.g. iron and silicon), and anions (F−, Cl−, Br−, NO, NO, SO, PO and organic acids (e.g. acetate, propionate, formate and butyrate) were measured. Further details on anion and cation analysis are provided in Sect. S2.
2.6 Air mass transport densities
To trace the origins of air masses arriving at the sampling site on Devon Ice Cap and characterize source regions, air mass back trajectories were computed using the hybrid single-particle Lagrangian integrated trajectory (HYSPLIT) model (Stein et al., 2015). Air parcel back trajectories were computed, allowing us to examine air mass moisture source regions and transport to the Devon Ice Cap. Back-trajectory analysis was performed using the National Centers for Environmental Prediction and Atmospheric Research (NCEP/NCAR) global atmospheric reanalysis dataset at 2.5∘ resolution. Daily, 10-day back trajectories were initiated at 2175 m a.m.s.l. at the Devon Ice Cap sample site location for years 1994 and 2013.
Residence time analysis was used to identify air mass transport densities for 1994 and 2013 (Miller et al., 2002). This approach analyzes a large number of trajectories to reduce uncertainties, develop reliable pathways of airflow and account for variations in transport speed and direction (Criscitiello et al., 2016). For this analysis, the total number of trajectory endpoints were summed within each equal-area pixel, and then divided by the zonal distance between the Devon ice core sampling site and each pixel to remove concentric patterning. The air mass transport densities were scaled on a 0–1 scale. In this study we focus on low-elevation air masses (0–500 m above terrain), which are more likely to be representative of evaporation moisture source.
3.1 PFAA concentrations and fluxes on the Devon Ice Cap: comparisons to previous studies
A comprehensive analysis of perfluoroalkyl acid (PFAA) and FOSA annual deposition on the Devon Ice Cap was carried out on ice core samples dating from 1977 to 2015. In general, PFCAs from TFA (C2) to PFTrDA (C13) were detected on the Devon Ice Cap. Deposition and temporal trends of TFA to PFBA (C4) will be discussed in a separate paper. Observed PFCA concentrations from PFPeA (C5) to PFTrDA ranged from < 3.21 to 755 pg L−1 (Fig. S1 in the Supplement, Table S9). PFCAs from PFHxA to PFUnDA (C6–C11) were detected in almost every sample, with the exception of PFUnDA, which was not detected in one year. The long-chain PFCAs, PFDoDA (C12), and PFTrDA were only quantifiable in < 3 years, while PFTeDA, PFHxDA, and PFOcDA were < LOQ throughout the 38-year time period and will not be discussed further. PFSAs including PFBS, PFHpS, and PFOS, as well as FOSA were detected on the Devon Ice Cap with concentrations ranging from < 0.18 to 391 pg L−1 (Table S10). PFOS was detected in every sample while PFBS and PFHpS were only detected > LOD in two years. Evidence of the presence of PFHxS, PFDS, and PFECHS was sought, but not found. FOSA was detected > LOD in most samples up until 2000 and only in three samples after 2000.
Concentrations (pg L−1) of the PFAAs were converted to fluxes (ng m−2 yr−1; Sect. S3) to determine annual deposition of these compounds in the Canadian Arctic. The dating of the ice core itself is discussed in more detail in the SI (Sect. S1). Annual snow accumulation was determined by measuring the length of the annual ice core sections. Annual snow accumulation was calculated as 0.15–0.64 m yr−1, which is consistent with 0.22–0.24 m wet equivalents yr−1 reported by (Pinglot et al., 2003) for the Devon Ice Cap from 1963 to 2000. Fluxes of PFAAs, including PFOA to PFUnDA (C8–C11) and PFOS (Tables S11, S12), were compared to fluxes of these PFAAs in two previous samples collected from snow pits on the Devon Ice Cap in 2006 (Young et al., 2007) and 2008 (MacInnis et al., 2017). The datasets generally agree within the uncertainty of the measurements, with the exception of PFOS (Figs. S2, S3). These slight discrepancies between the Devon Ice Cap studies could be the result of multiple factors. In the 2006 study, there was limited availability of isotopically labelled and native standards of sufficient purity. Our study represents an improvement in analytical methods since that time, both in terms of instrument detection limits and accuracy. The 2006 and 2008 sampling strategies are also in contrast to the current approach of ice core drilling. In the earlier efforts, depth samples were obtained by horizontal cylindrical sampling the face of an ice pit (2006) and vertically sampling the face of a snow pit continuously (2008). Those datasets represent semi-continuous depth measurements. In our current method, we obtained ice cores using a custom drill designed for Arctic sampling and conventional practices in ice sampling for temporal profiling (Boon et al., 2010; Readinger, 2006). Lastly, the Devon Ice Cap sampling locations in the earlier research were not at the summit of the ice cap as in the current research. These differences in location may have resulted in some variability in fluxes. The most straightforward temporal record of atmospheric deposition is obtained at the summit of an ice cap (Legrand and Mayewski, 1997); thus, the current research is hypothesized to be a better representation of PFAA deposition to Devon Island.
Concentrations and homologue trends of PFCAs and PFSAs detected in this study are comparable to several other studies (Table S13). Comparable levels of PFCAs ranging from PFPeA to PFUnDA were detected in remote snow core and surface snow samples from Cole Gnifetti (Kirchgeorg et al., 2013), Longyearbyen (Kwok et al., 2013), and Livingston Island (Casal et al., 2017); and from glaciers on the Tibetan Plateau and Lake Namco (X. Wang et al., 2014). Concentrations of the PFCAs were generally much lower in the Arctic and Atlantic Oceans compared to the Devon Ice Cap concentrations. PFOS and FOSA concentrations were variable among all studies with no discernable trend for sample matrix or sample site.
3.2 Air mass transport density analysis
Previous studies using back trajectory analysis of air masses (Kahl et al., 1997; Meyer et al., 2012) have provided evidence for atmospheric LRT to the Devon Ice Cap from North America and Eurasia. Air masses on Devon Island originated three times more often from populated regions of Northern Europe and Asia compared to North America, and southern and eastern Asia were significant source regions. Little variation was observed in Devon Ice Cap air mass source regions over the time period 1994–2008 (Meyer et al., 2012). Previous studies on spatial patterns of pollen deposition in the High Arctic further support these findings. The Devon Ice Cap is located within an air mass boundary between 74 and 76∘ N, between sites north of 76∘ that receive dominantly Eurasian pollen sources and sites south of 74∘ that receive dominantly North American pollen sources (Bourgeois et al., 2001). Devon Ice Cap therefore receives air masses and pollen/pollutant sources from both North America and Eurasia.
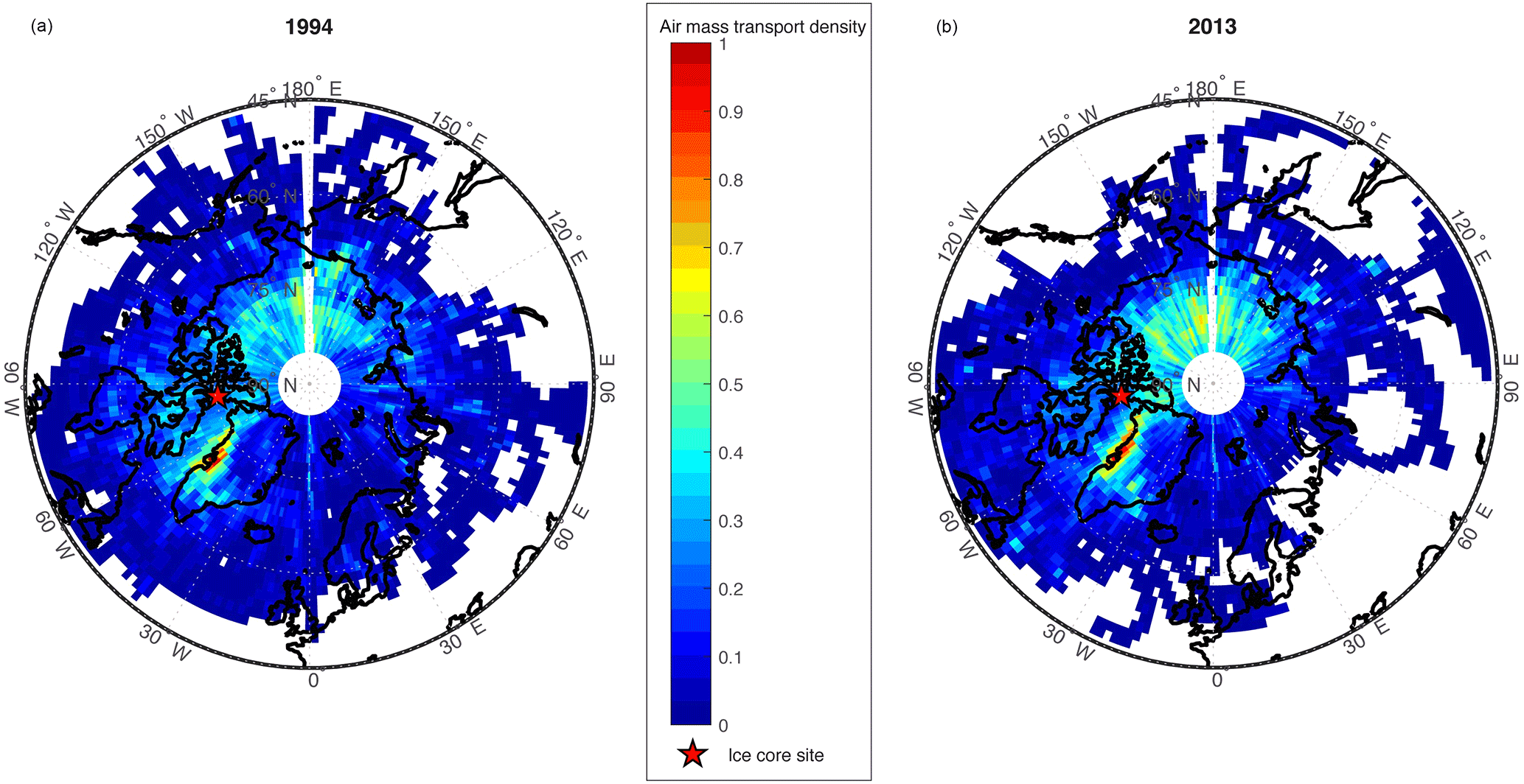
Figure 1Air mass transport density maps (scaled 0–1) for air parcels reaching the Devon Ice Cap ice core site (red star), for (a) 1994 and (b) 2013.
Air mass transport densities have previously been employed for investigating probable source regions and flow pathways of air masses in the Canadian Arctic (Criscitiello et al., 2016). Air mass transport density analyses calculated using the HYSPLIT model for the Devon Ice Cap for the years 1994 and 2013 are shown in Fig. 1. The year 1994 was selected to compare to previous trajectory analyses conducted by Meyer et al. (2012), and showed similar results. Due to the high PFOS flux observed in 2013, we also examined this year to identify any transport anomalies, but found both years (1994 and 2013) to be comparable. Air mass transport densities for both 1994 and 2013 reveal elevated residence time densities in parts of Asia, and particularly high residence time densities along the west coast of Greenland.
3.3 PFCA deposition and temporal trends on the Devon Ice Cap
Annual fluxes of PFCAs ranged from < LOD to 141 ng m−2 yr−1 (Table S11). Temporal trends of shorter-chain PFCAs (C2–C4) will be discussed separately, so this section will focus solely on PFCAs with at least five carbons. In general, the flux of PFCAs increased after 1985 (Fig. S4), but with diverging trends from 1995 to 2015. Various glaciology studies have shown that the Devon Ice Cap has experienced strong summer warming since 2000 and especially after 2005 (Bezeau et al., 2013; Gascon et al., 2013; Sharp et al., 2011). This is consistent with Inuit traditional knowledge of overall warming in the Arctic (Eetoolook et al., 2001). The variability in fluxes within the past 15–20 years could, therefore, be partially attributed to melting effects. When melting events occur, the ability to discern temporal trends in chemical deposition of various compounds can be compromised by the percolation of meltwater or elution of particles by meltwater flow (Eichler et al., 2001; Steinlin et al., 2016). These melting events could bias annual flux measurements of PFAAs in ice core samples during the melt period, since PFAAs on the ice cap surface can be eluted into the snowpack, before refreezing at ice layer interfaces where temperatures at depth are below the pressure melting point (Bezeau et al., 2013). One study has examined elution behaviour of PFAAs from a melting snowpack and found that elution of PFAAs is driven by a number of factors including water solubility in the snowpack, PFAA chain length, and sorptive capacity of the snow grain surface (Plassmann et al., 2011). Due to melting at the surface, concentrations of PFAAs measured near the surface layer can result in inaccurate estimations whereby one year might be overestimated and another year underestimated. Melting events on the ice cap have likely happened periodically over the last 15–20 years, thereby blurring the vertical profile to some extent. However, we expect any melting that occurred to have primarily affected the seasonal trends, as Koerner (2005) states that the percolation of meltwater in a snowpack will refreeze within an annual layer, and so seasonal cycles of PFAA deposition will be biased, but annual interpretations should not be affected (Koerner, 2005). Variability of ±1 year could be caused by inaccuracies in dating and/or error associated with ice core sectioning. To circumvent the compounding impacts of recent melt events and consequent meltwater percolation, and any error associated with ice core dating and sectioning, a 5-year moving average was applied to the flux measurements, thereby facilitating long-term temporal trend analysis of PFAS deposition to Devon Ice Cap.
Both PFOA and PFNA fluxes increased from 1977 up until at least 1995 (Fig. 2). In the period post-1995 until 2013, fluxes have plateaued, with < 25 ng m−2 yr−1 variance in annual flux. From 2012 to 2015, a large decline in PFOA and PFNA fluxes is apparent. The decrease in flux post-2012 was noted for the entire suite of PFCAs (Fig. S4). The most recent decrease in PFCA fluxes could be due to melting events or ice core dating and sectioning inaccuracies, but it also corresponds to anticipated PFCA emission reductions through the United States Environmental Protection Agency (EPA) PFOA Stewardship Program (US EPA, 2016), as well as the Canadian Environmental Performance Agreement (Environment and Climate Change Canada, 2006). In 2006, the EPA invited eight major fluoropolymer and fluorotelomer manufacturers to commit to eliminating emissions and product content levels of PFOA, precursor compounds, and related longer chain length homologue chemicals. Corporations voluntarily committed to achieving a 95 % reduction by 2010, measured from a year 2000 baseline, and full elimination of these products and emissions by 2015 (US EPA, 2016). In Canada, the federal government established The Environmental Performance Agreement with the same commitment between Environment and Climate Change Canada (ECCC), Health Canada and four major manufacturers with known organofluorine products in Canadian commerce (Environment and Climate Change Canada, 2006). The most recent data for company-reported reductions in emission and product content for the US, Canada, and non-US operations are summarized in Tables S14–S15. As of 2016, all companies participating in the PFOA Stewardship Program and Environmental Performance Agreement reported they had met the goals of the program. As part of both agreements, all major manufacturers reduced their production and emissions of PFOA and related compounds by at least 95 % from 2006 to 2010. The observed decrease in PFOA and PFNA fluxes from 2012 to 2015 cannot solely be attributed to these phase-outs, since it would be expected that the phase out would cause a large decrease in PFCA deposition between 2006 and 2010 and a small decrease after 2012. It is probable that existing products continued to emit after the stewardship program took effect, which could delay the detection of its impact (Prevedouros et al., 2006). This is evident in temporal trend analysis in Canada and the US such as in human serum, freshwater fish, and non-migratory birds, which do not show much of a decline in PFCAs from 1990s to 2012 (Braune and Letcher, 2013; Calafat et al., 2007; Gewurtz et al., 2016; Olsen et al., 2003).
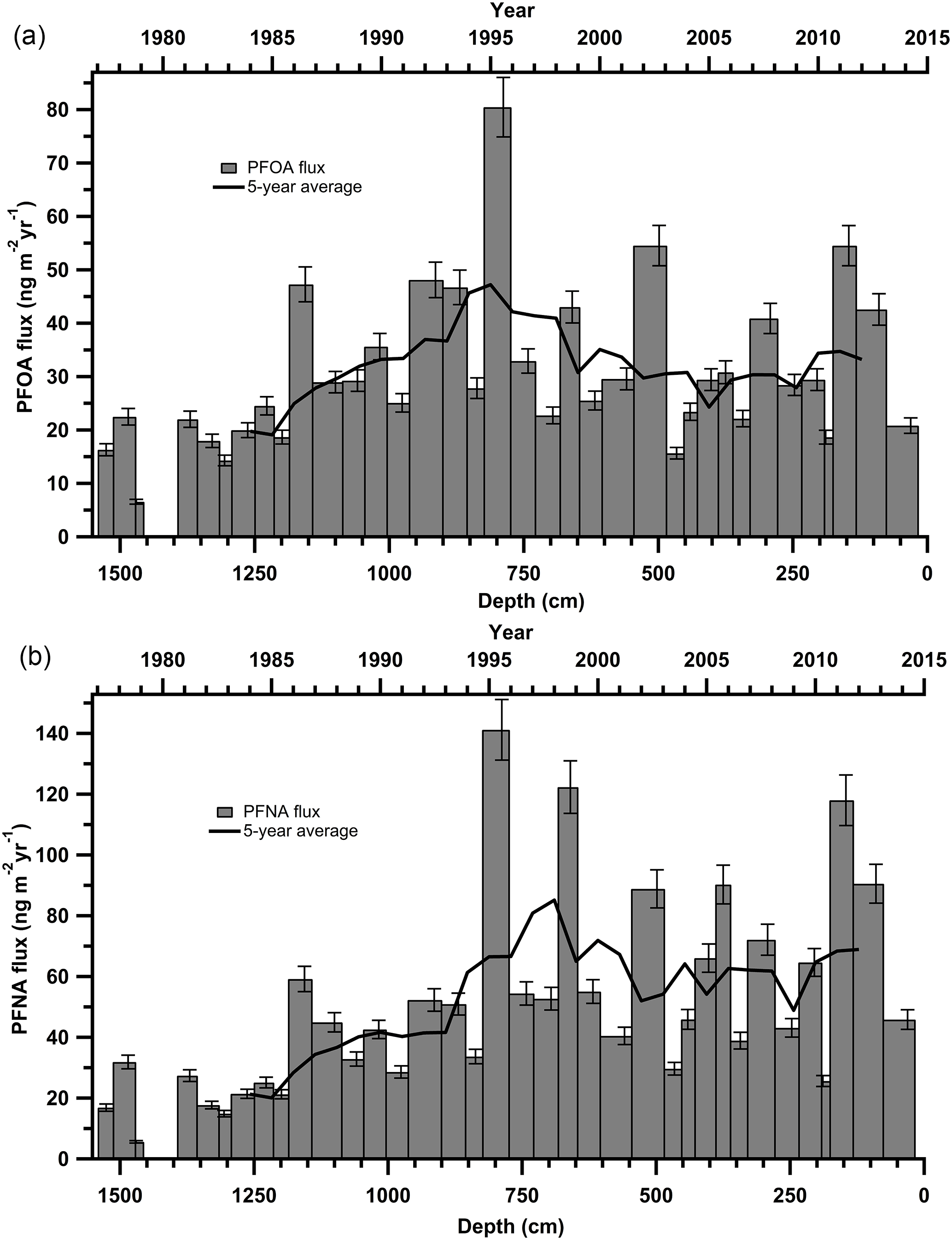
Figure 2Annual deposition fluxes on the Devon Ice Cap: (a) PFOA and (b) PFNA. The solid black line represents the 5-year moving average.
In addition, other historical and on-going sources of PFASs have emerged. Manufacturers that were not signatories to the PFOA stewardship program have been producing PFASs since 1985. China started polytetrafluoroethylene (PTFE) production around 1985 and was producing up to 60 kt of PTFE in 2015 (Wang et al., 2014a). Some manufacturers have emerged more recently that produce PFAAs and precursor compounds (Land et al., 2015; Wang et al., 2014a). In general, from 2000 onward, long-chain (> C8) PFCAs have been phased-out through regulation or voluntary reduction by major producers in Japan, Western Europe, the United States and Canada (Wang et al., 2014a, b). Meanwhile, new manufacturers (e.g. China) have begun producing these long-chain PFCAs and their precursors (Land et al., 2015). The total estimated annual emissions of PFBA to PFTeDA for Canada, the United States, Western Europe, and Japan were 25–50 t yr−1 in 2010, while estimated emissions of PFBA to PFTeDA were 40–193 t yr−1 in China in 2013 (Wang et al., 2014a). Thus, global emissions of PFAAs and their precursors have not decreased significantly, which is consistent with observed temporal trends on the Devon Ice Cap. Furthermore, the phase-out has created a market for numerous alternative fluorinated products (Wang et al., 2016, 2013), some of which may be precursors to these long-chain PFCAs. Observed temporal trends in PFAA fluxes may represent combined effects of the stewardship program in North America and increasing production and emissions in other regions.
3.4 PFSA and FOSA deposition and temporal trends on the Devon Ice Cap
The observed concentrations of PFSAs and FOSA correspond to annual fluxes from < LOD to 80.3 ng m−2 yr−1 (Table S12). Since PFBS and PFHpS were only detected in three samples, there are no observable trends. PFOS and FOSA each show distinct temporal trends. PFOS was detected at consistent levels below 10 ng m−2 yr−1, with an anomaly (80 ng m−2 yr−1) detected in 2013 (Fig. 3a). FOSA was measured in almost every year from 1977 to 2000 with fluxes increasing until 1995. After 2000, FOSA was only detected in three samples with levels < 0.76 ng m−2 yr−1 (Fig. 3b). FOSA is a known volatile precursor and can degrade to PFOS (D'eon et al., 2006); however, PFOS was continually measured after 2000, whereas FOSA was not. In addition, there was no correlation between PFOS and FOSA measurements (Table S16). Therefore, it is reasonable to assume that there are sources of PFOS that are independent of FOSA and other sulfonamido derivatives.
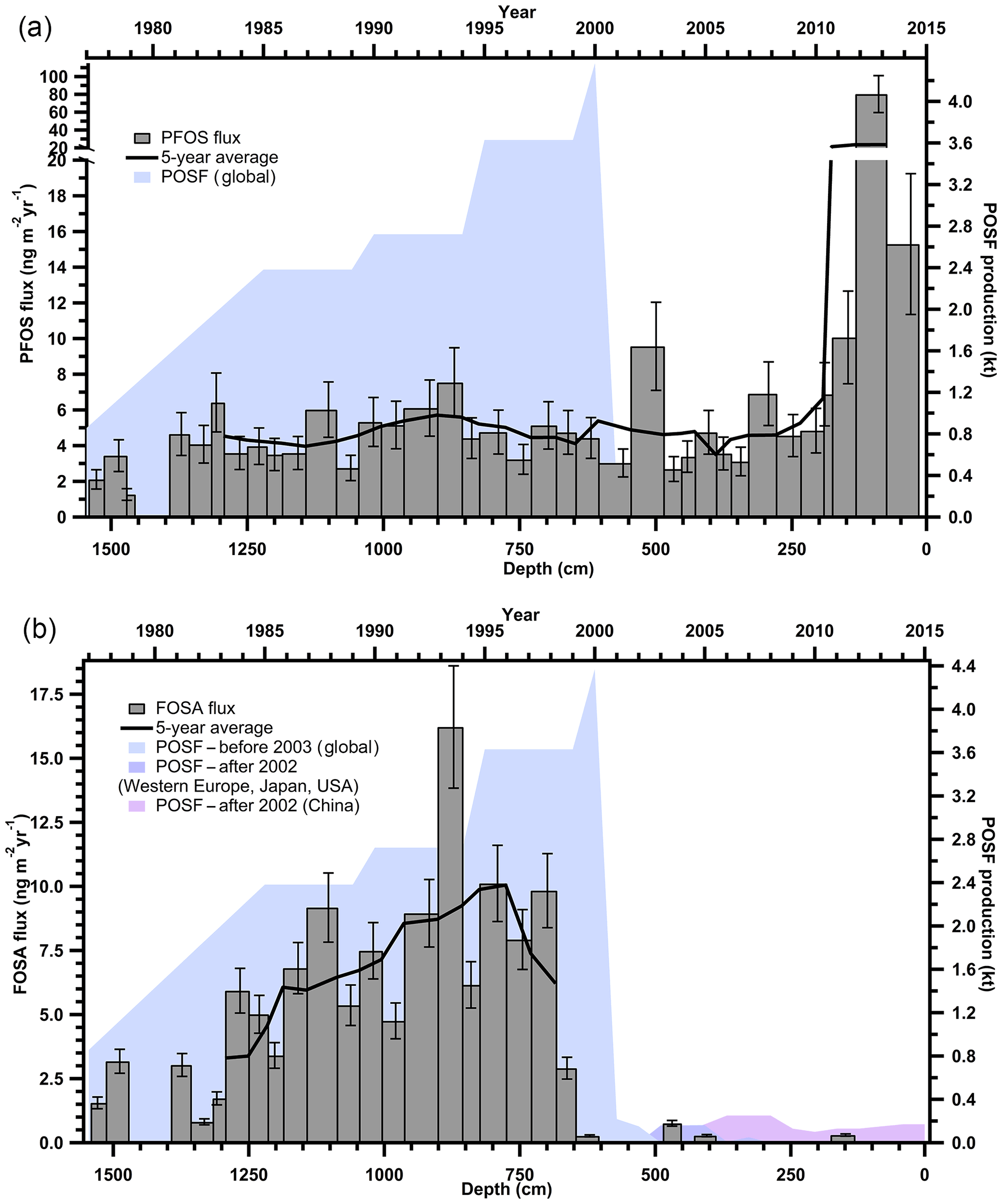
Figure 3Annual deposition fluxes on the Devon Ice Cap: (a) PFOS and (b) FOSA, with global POSF production from 1977 to 2003 and POSF production for Western Europe, Japan, United States and China from 2003 to 2015 (Wang et al., 2014a, 2017). Solid black lines represent 5-year moving averages.
From 2000 to 2002, 3 M, one of the major global producers of perfluoroalkyl substances phased out the production of the synthetic precursor to FOSA and PFOS, perfluorooctane sulfonyl fluoride (POSF), as well as related products based on C6, C8, and C10 chemistry (Wang et al., 2013; Weppner, 2000). These were replaced with C4-based chemistry, where products were derived from perfluorobutane sulfonyl fluoride (PBSF). These compounds are believed to have lower bioaccumulative and toxicological effects (Stahl et al., 2011). Reported production of PBSF in the United States almost doubled from 2002 to 2006, while that of POSF decreased by more than two orders of magnitude between 1998 and 2002, with no known production after 2006 (Wang et al., 2014b). This is interesting because we detected quantifiable levels of PFBS in the ice core in two recent years, and so our understanding of LRT of these PFASs is incomplete and warrants further research. Before 2003, 3M was responsible for most global PFOS production (Paul et al., 2009). By 2006, PFOS and related substances derived from POSF were regulated under the European Union (EU) Directive 2006/122/EC and by 2009 were listed under Annex B (restriction of production and use) of the Stockholm Convention on Persistent Organic Pollutants coordinated by the United Nations Environment Programme (UNEP; Land et al., 2015). These production changes in PFOS in the early 2000s were used to explain temporal trends in the Canadian Arctic (Butt et al., 2007). The decline of precursor POSF production, and hence of all C8 sulfonamido derivatives, including FOSA, by 3M is consistent with FOSA trends observed on the Devon Ice Cap, in which the majority of FOSA is < LOD after 2000, suggesting an effective phase out of this compound (Fig. 3b). This was similarly observed in samples collected from Devon Ice Cap in 2008 (MacInnis et al., 2017), in North Atlantic pilot whales harvested between 1986 and 2013 (Dassuncao et al., 2017), and in Arctic air at Alert (Hung et al., 2016).
Temporal trends of PFOS deposition to the Devon Ice Cap do not reflect the production phase out of PFOS by 3M in the early 2000s (Fig. 3a). Rather, PFOS production and manufacturing, along with replacements (e.g. PFBS), have increased dramatically in Asia since 2001, and China is now the dominant producer of these compounds (Lam et al., 2016; Xie et al., 2013; Yao et al., 2016). PFOS production in China began increasing rapidly around 2000 and is currently steady at 100–200 t yr−1 (Wang et al., 2016). China was reported to be the main producer and user of PFOS substances between 2003 and 2008 with less than 50 t in 2003 and up to 250 t of POSF-based products produced in 2008 (Lim et al., 2011). During this time, over 100 t of PFOS was also used annually in China to produce aqueous film forming foams (AFFFs; Armitage et al., 2009b) used for extinguishing fuel-based fires. According to available 2006 inventories, 15 Chinese enterprises were producing over 200 t of POSF, of which 100 t were for export (Ruisheng, 2008). This suggests the annual volume of PFOS production in China in the mid-2000s was similar to the annual production by 3 M in the late 1990s (Armitage et al., 2009b). Although it is known that the production of perfluoroalkane sulfonyl fluorides has increased in China, global emission data for individual compounds are currently unavailable and cannot be correlated with the temporal trends observed on the Devon Ice Cap (Lim et al., 2011). However, the continuous detection of PFOS after the early 2000s on the Devon Ice Cap may be related to the on-going production and use of PFOS substances by manufacturers in Asia (Wang et al., 2017). Production of other PFOS-related perfluorinated chemicals is on-going in China, as well as in Russia and India (Jiang et al., 2015) which is supported by higher levels of PFOS after 2011 in the Devon Ice Cap. An anomalously high PFOS flux was observed in 2013. This flux was between five and eight times greater than both previous and following years. There was no signal enhancement for other PFASs in 2013 and the air mass transport model showed no transport anomaly for this year. Therefore, the large 2013 PFOS flux in the Devon Ice Cap is unlikely to be attributed to contamination during method collection or analysis, or due to air mass movements. The reason for the high 2013 PFOS flux is unknown but may suggest other sources. Arctic air samples collected at Alert also showed the highest levels of PFOS in 2013 (Hung et al., 2016), and anomalously high PFOS levels were observed in landlocked Arctic Char from Cape Bounty, Melville Island in Nunavut, Canada, collected between 2011 and 2015 (Cabrerizo et al., 2016). This increasing PFOS trend warrants further consideration in order to determine the efficacy of current POSF restrictions.
3.5 PFCA homologues and volatile precursors
Indirect sources of PFAAs are contributors to the global presence of these compounds, particularly in locations such as the Canadian Archipelago (Benskin et al., 2012a). This has been demonstrated in multiple studies that detected the presence of volatile precursors (e.g. FTOHs, (N-Alkyl) FASAs/FASEs), and FTOH-precursor degradation products (e.g. fluorotelomer unsaturated carboxylic acids (FTUCAs)), in the Canadian Arctic (Benskin et al., 2011; Schenker et al., 2008; Shoeib et al., 2006; Stock et al., 2007). Volatile precursor compounds will oxidize in the atmosphere to produce PFCAs. Patterns of PFCA homologues are useful in examining the role that fluorotelomer-derived compounds play in gas-phase atmospheric oxidation (Young et al., 2007). If these compounds are coming from the same source, then sequential pair concentrations are expected to vary through time together. There will be some variability in the ratios depending on the relative atmospheric levels of NOx (NO + NO2) and peroxy radicals (Young and Mabury, 2010). In this study, comparisons were made between observed concentrations of 36 pairs of PFCA homologues ranging from PFPeA to PFTrDA (Table S16). Most sequential pairs of PFCA homologues were deposited in similar amounts on the ice cap. Correlations between sequential pairs from PFPeA and PFDoDA were all statistically significant (two-tailed t-test) with strong correlations (all p-values ≤ 0.0001; 0.527 ≤ R2≤ 0.889; Table S16). Similar correlations were observed for a number of PFCA homologues in previous ice samples from the Devon Ice Cap (MacInnis et al., 2017) and from Longyearbyen, Svalbard (Kwok et al., 2013). The correlations are consistent with expected PFCA homologue production via gas phase atmospheric oxidation of fluorotelomer-derived compounds (Ellis et al., 2004). Flux measurement ratios were calculated for six pairs of PFCA homologues from PFPeA to PFUnDA over the time series (Fig. S5). The three major even–odd pairs expected to be formed from 6:2, 8:2, and 10:2 fluorotelomer compounds are PFHxA : PFHpA, PFOA : PFNA, and PFDA : PFUnDA, respectively. The majority (82 %) of the flux ratio measurements were within a factor of two, supporting the hypothesis that these sequential, even–odd homologues are likely coming from fluorotelomer-derived sources (Fig. 4a; Ellis et al., 2004; Wallington et al., 2006). Fluorotelomer compounds of different chain lengths were produced and used to different extents. We can compare these using odd–odd PFCA homologue ratios, comparing PFNA to PFPeA, PFHpA, and PFUnDA, as products of 8:2, 4:2, 6:2, and 10:2 fluorotelomer compounds, respectively (Fig. 4b). The dominant homologues are PFNA and PFHpA, followed by PFPeA, then PFUnDA. This suggests that 8:2 and 6:2 fluorotelomer compounds dominate as precursors, followed by the 4:2 and 10:2 fluorotelomer compounds. This trend is consistent with our knowledge of commercial product formulations and atmospheric measurements (Dinglasan-Panlilio and Mabury, 2006; Heydebreck et al., 2016; Young and Mabury, 2010). Despite producers moving from 8:2 to shorter-chain formulations, recent FTOH atmospheric measurements have found that the 8:2 FTOH remains the dominant compound in the high European Alps (Xu et al., 2017). Therefore, it is likely that PFCAs from PFPeA to PFUnDA on the Devon Ice Cap are derived from common emission sources due to prominent quantities of residual volatile precursors in fluoropolymer products.
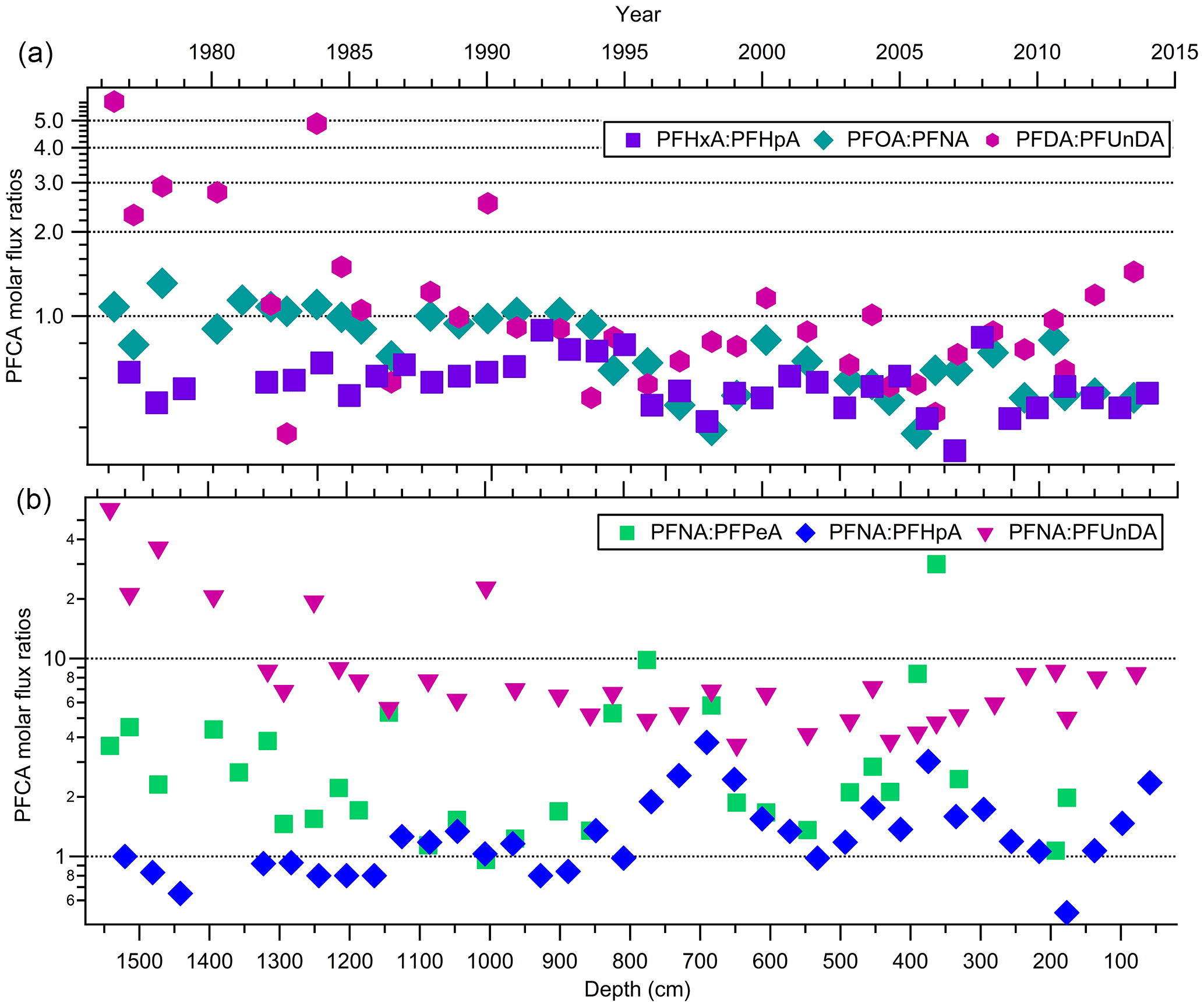
Figure 4Molar flux ratios for (a) three even–odd pairs of PFCAs and (b) three odd pairs of PFNA with PFPeA, PFHpA, and PFUnDA homologues, as a function of depth and year.
The atmospheric oxidation of perfluoroalkane sulfonamido substances may provide an additional source of PFCAs to the Devon Ice Cap. The oxidation of FOSA could contribute to the observed flux of PFOA and shorter-chain PFCAs in the High Arctic (Martin et al., 2006). However, since there were no observed correlations between FOSA and PFOA or FOSA and any of the other PFCAs (p≥ 0.0021; R2 ≤ 0.24, Table S16), it is likely that PFOA deposition to the Devon Ice Cap is driven by other sources.
3.6 Elucidating the role of marine-driven transport of PFAAs to the Arctic
Thus far, evidence supports indirect formation of PFAAs in the atmosphere from volatile precursor compounds as the major source, indicating that direct transport is likely a minor source of PFAAs to the ice cap. Atmospherically formed PFAAs can deposit to any terrestrial or oceanic system. Those PFAAs deposited to oceans can re-enter the atmosphere in the form of marine aerosols (McMurdo et al., 2008). It is therefore important to understand the extent to which the oceans contribute to PFAA ice cap deposition. Other sources, such as dust or biomass burning, must also be considered as possible contributors of PFAAs to the ice cap. Gas-phase PFAAs will deposit through wet or dry deposition and PFAAs may also be transported on aerosols (Thackray and Selin, 2017). Major ions are useful source markers for atmospheric aerosols and were measured in the ice core to further understand these transport mechanisms (Tables S17–S18, Figs. S6–S7).
The non-sea salt (nss) component of the ice core samples was calculated to understand the atmospheric origin in the samples (Keene et al., 1986). All sodium (Na+) in the ice core samples was assumed to come from sea salt. Most other ions were attributed to the nss component, suggesting limited oceanic sources depositing on the Devon Ice Cap (Table S19). Further, no correlations were observed between Na+ flux and any of the PFAAs (p≥ 0.0093; R2≤ 0.218; Table S20).
Another technique used to assess the influence of marine aerosol deposition of PFAAs to the Devon Ice Cap is a comparison between ocean and ice cap homologue patterns (MacInnis et al., 2017). If marine aerosols were/are a major source of PFAA contamination on the Devon Ice Cap, then one would expect the homologue profiles to be similar between the ocean and the ice cap. Water-to-air transport of PFAS is related to surfactant strength (Reth et al., 2011). Consistent with results from MacInnis et al. (2017), differences in surfactant strength cannot account for the different homologue profiles observed on the ice cap and in the ocean (Fig. S8). Figure 5 illustrates the proportional analysis of the molar concentration (pmol L−1) fraction between PFAAs on the Devon Ice Cap and ocean levels in the Canadian Arctic Archipelago (Benskin et al., 2012b), Arctic Ocean (Benskin et al., 2012b; Cai et al., 2012a), North Atlantic Ocean (Zhao et al., 2012), and North Pacific Ocean (Cai et al., 2012a) for the years 2005 and 2010. Molar concentration fractions of PFAAs on the ice cap differed from ocean PFAA fractions, with higher molar concentration fractions of PFBA and PFNA, and lower molar concentration fractions of PFSAs found on the Devon Ice Cap. PFHxS was not detected on the ice cap but was measured in most ocean samples. As described by MacInnis et al. (2017), PFHxS can act as a marker for direct transport. The absence of PFHxS on the ice cap is further confirmation that marine aerosols are not a significant source for PFAA deposition to the Arctic environment. The differences in homologue profiles between the ice cap and the ocean can also not be accounted for by different surfactant properties (MacInnis et al., 2017), suggesting the two may have different sources of PFAA contamination. Additionally, correlations between annual PFAS deposition and Arctic sea ice minimum were calculated (NSIDC, 2017; Table S22). If marine aerosols were a major source of PFAS to the ice cap, then a negative correlation between annual sea ice extent and deposition would be expected, but the majority (12 of 14) showed a positive correlation. The discrepancies between the Na+ ∕ PFAA flux ratios and the ice cap/ocean proportional analysis provide further evidence to imply that marine aerosols are not a significant source of PFAAs to the Devon Ice Cap.
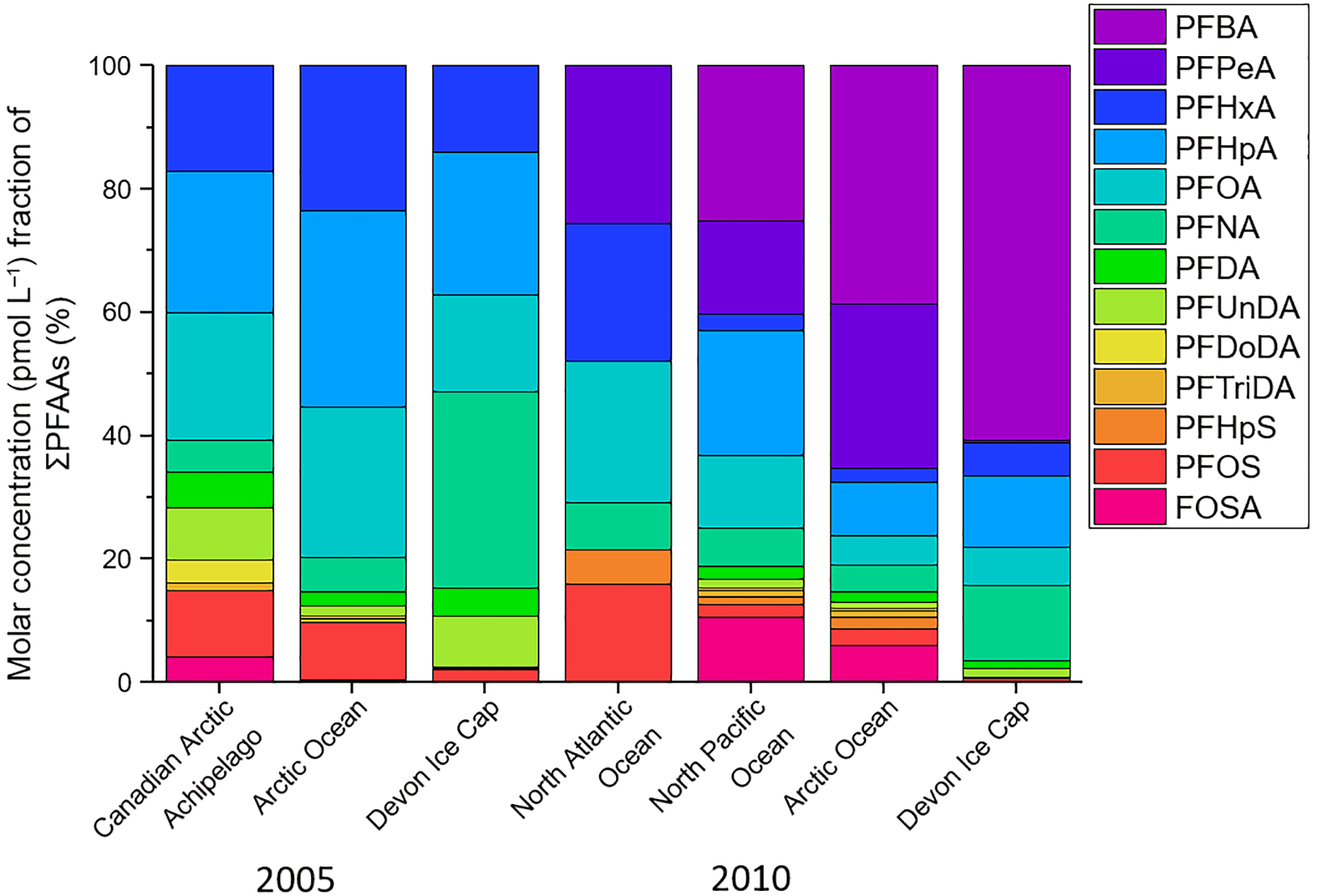
Figure 5Molar concentration fraction of sum of PFAAs on the Devon Ice Cap (from the current 2015 ice core collection) compared to levels in the Canadian Arctic Archipelago (Benskin et al., 2012b), Arctic Ocean (Benskin et al., 2012b; Cai et al., 2012a), North Atlantic Ocean (Zhao et al., 2012), and North Pacific Ocean (Cai et al., 2012a) in 2005 and 2010. Ocean concentrations are compared to ice cap concentrations for two years to show a better overall comparison with different ocean samples that were collected in multiple field campaigns.
3.7 Understanding atmospheric inputs of PFAAs using ion tracers
Weak correlations were observed between short-chain PFCAs (C2–C4) and nss-flouride (F−) (p≤ 0.0015; 0.306 ≤ R2≤ 0.455). A small percentage of the F− being detected in the ice core samples may be derived from the atmospheric formation of PFCAs. The degradation of many PFCA precursors forms both PFCA and HF. For example, the hydrolysis of perfluoroacyl fluorides forms the corresponding PFCAs along with the loss of HF (Calvert et al., 2008).
By this mechanism in Eq. (1), PFCAs could account for between 0.80 and 14 % of the F− present on the ice cap, depending on the year. These numbers are upper limits as PFCAs can also be formed by mechanisms that do not form HF (Young and Mabury, 2010). It is difficult to assess the exact contribution of this anthropogenic source to the overall burden of F−, due to the lack of available data on F− sources. This is further confounded by the high mobility of F− in both firn and ice layers, which makes it difficult to study temporal trends in F− deposition (Preunkert et al., 2001). Both natural and anthropogenic sources contribute to the overall budget of F− in the troposphere, including primary sea-salt, soil dust aerosols, volcanic emissions, coal burning, and industrial processing (Preunkert et al., 2001). We are currently unable to assess the exact contributions of each source of F−, but the correlations observed here suggest that short-chain PFCA precursor degradation could account for up to 14 % of the observed F−. Furthermore, these correlations lend further support to indirect formation as a major pathway to PFCA contamination on the Devon Ice Cap. The role of ice in the formation of PFAAs from precursors is currently unknown and future work should also focus on ice-mediated PFAA formation.
There were weak to moderate correlations between several PFAAs and nss-Ca2+ and nss-Mg2+ (0.300 ≤ R2≤ 0.549), both of which are indicators of mineral dust (Mochizuki et al., 2016). Mineral dust aerosols can accumulate acidic atmospheric contaminants, such as nitric and hydrochloric acids (Sullivan et al., 2007). Dust can undergo LRT and deposit in the remote Arctic (Rahn et al., 1977; Zdanowicz et al., 1998). Dust entrainment in deserts is one of the most important sources of mineral dust in the global atmosphere and North African (e.g. Sahara) and Central Asian (e.g. Gobi desert) dust sources contribute the most global dust to the Northern Hemisphere (Luo et al., 2003). Several studies have found dust particles associated with LRT in snow and ice samples from the Canadian Arctic (Bullard et al., 2016; Groot Zwaaftink et al., 2016; Zdanowicz et al., 1998). Dust deposition to the Arctic shows a seasonal effect, with dust storms in major deserts occurring more frequently in the spring, leading to higher concentrations of mineral dust tracers in the spring and autumn (Barrie and Barrie, 1990). We detected high concentrations (199–786 µg L−1) of Ca2+ on the Devon Ice Cap, and calculated that the majority (99.5 %) of Ca2+ contributed to the nss component. Substantial concentrations (< 2.00–78.3 µg L−1) of other mineral dust (Al3+, Fe3+, Si4+) tracers were also detected. Correlations between nss-Ca2+ and PFAAs suggest there is a relationship between the transport of mineral dust and PFAAs to the Devon Ice Cap. This could be caused by both mineral dust and PFAAs originating from the same regions. Alternatively, it could indicate a mechanistic relationship. Atmospheric acids are known to interact with mineral dust through reactive uptake (Sullivan et al., 2007). It is possible that PFAAs, as strong atmospheric acids, could behave in the same way and be taken up onto mineral dust aerosols and subsequently transported to the Arctic. We cannot distinguish between these mechanisms at this time and suggest that further studies explore this relationship between mineral dust and PFAA LRT.
Monitoring of temporal trends in persistent organic pollutant deposition in remote areas is an important initiative to determine the impact of regulation on contamination of pristine environments. This study demonstrates the value of ice cores to understanding contaminant LRT. Herein we report the first multi-decadal record of PFAA deposition in the Canadian Arctic. Continuous and increasing deposition of many PFAAs on the Devon Ice Cap was observed, suggesting on-going emission and use of these PFAAs and their precursors in areas such as North America and Eurasia. These results indicate that the Devon Ice Cap is likely impacted by global pollutants from both North American and Eurasian Sources, with Continental Asia becoming a greater contributor in recent years. Use of major ion tracers provided new information regarding the transport of PFAAs in the atmosphere, confirming that marine aerosol inputs are unimportant and suggest a relationship with mineral dust. We also observed that a small percentage of fluoride detected in the Arctic could be coming from the reactive mechanisms forming the PFAAs. Further efforts are necessary to continue monitoring the long-range transport of PFAAs and their deposition to the remote Canadian Arctic. It is important to understand these LRT mechanisms and determine the geographical sources of PFAAs and their precursors. Ice cores can aid in elucidating these mechanisms and further collection and analysis of ice cores is recommended.
All data is included in the Supplement.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-5045-2018-supplement.
HMP sectioned, extracted and analyzed the ice core, conducted the data analysis, and wrote the manuscript. CJY designed and led the project, directed ice core sectioning, assisted with data analysis, and edited the manuscript. ADS assisted in ice core analysis, data interpretation, and edited the manuscript. CS aided in ice core extraction and conducted analysis. ASC conducted transport modelling and ASC and MJS coordinated field operations, collected the ice core, and performed the dating of the ice core. DCGM provided input on the manuscript.
The authors declare that they have no conflict of interest.
This work was funded by the Northern Contaminants Program (Indigenous and
Northern Affairs Canada) to Cora J. Young, Amila O. De Silva, Derek C. G. Muir, and Martin J. Sharp, and Natural
Resources Canada (Polar Continental Shelf Project) to Alison S. Criscitiello. We also
acknowledge NSERC Discovery Grants to Cora J. Young and Martin J. Sharp and Northern
Research Supplement to Martin J. Sharp. We thank Colleen Mortimer and Anja Rutishauser
for assistance with ice core collection; John MacInnis and Cyril Cook for
assistance with ice core sectioning; and Trevor VandenBoer and Jamie Warren
for assistance with ion chromatography analysis.
Edited by: John Liggio
Reviewed by: James Franklin and one anonymous referee
Armitage, J. M., MacLeod, M., and Cousins, I. T.: Comparative Assessment of the Global Fate and Transport Pathways of Long-Chain Perfluorocarboxylic Acids (PFCAs) and Perfluorocarboxylates (PFCs) Emitted from Direct Sources, Environ. Sci. Technol., 43, 5830–5836, https://doi.org/10.1021/es900753y, 2009a.
Armitage, J. M., Schenker, U., Scheringer, M., Martin, J. W., MacLeod, M., and Cousins, I. T.: Modeling the Global Fate and Transport of Perfluorooctane Sulfonate (PFOS) and Precursor Compounds in Relation to Temporal Trends in Wildlife Exposure, Environ. Sci. Technol., 43, 9274–9280, https://doi.org/10.1021/es901448p, 2009b.
Barrie, L. A. and Barrie, M. J.: Chemical Components of Lower Tropospheric Aerosols in the High Arctic?: Six Years of Observations, J. Atmos. Chem., 11, 211–226, https://doi.org/10.1007/BF00118349, 1990.
Benskin, J. P., Phillips, V., St. Louis, V. L., and Martin, J. W.: Source Elucidation of Perfluorinated Carboxylic Acids in Remote Alpine Lake Sediment Cores, Environ. Sci. Technol., 45, 7188–7194, https://doi.org/10.1021/es2011176, 2011.
Benskin, J. P., Ahrens, L., Muir, D. C. G., Scott, B. F., Spencer, C., Rosenberg, B., Tomy, G., Kylin, H., Lohmann, R., and Martin, J. W.: Manufacturing origin of perfluorooctanoate (PFOA) in Atlantic and Canadian Arctic seawater, Environ. Sci. Technol., 46, 677–685, https://doi.org/10.1021/es202958p, 2012a.
Benskin, J. P., Muir, D. C. G., Scott, B. F., Spencer, C., De Silva, A. O., Kylin, H., Martin, J. W., Morris, A., Lohmann, R., Tomy, G., Rosenberg, B., Taniyasu, S., and Yamashita, N.: Perfluoroalkyl Acids in the Atlantic and Canadian Arctic Oceans, Environ. Sci. Technol., 46, 5815–5823, https://doi.org/10.1021/es300578x, 2012b.
Bezeau, P., Sharp, M., Burgess, D., and Gascon, G.: Firn profile changes in response to extreme 21st-century melting at Devon Ice Cap, Nunavut, Canada, J. Glaciol., 59, 981–991, https://doi.org/10.3189/2013JoG12J208, 2013.
Boon, S., Burgess, D. O., Koerner, R. M., and Sharp, M. J.: Forty-seven years of research on the Devon Island Ice Cap, Arctic Canada, Arctic, 63, 13–29, https://doi.org/10.14430/arctic643, 2010.
Bourgeois, J. C., Gajewski, K., and Koerner, R. M.: Spatial patterns of pollen deposition in arctic snow, J. Geophys. Res.-Atmos., 106, 5255–5265, https://doi.org/10.1029/2000JD900708, 2001.
Braune, B. M. and Letcher, R. J.: Perfluorinated Sulfonate and Carboxylate Compounds in Eggs of Seabirds Breeding in the Canadian Arctic: Temporal Trends (1975–2011) and Interspecies Comparison, Environ. Sci. Technol., 47, 616–624, https://doi.org/10.1021/es303733d, 2013.
Buck, R. C., Franklin, J., Berger, U., Conder, J. M., Cousins, I. T., de Voogt, P., Jensen, A. A., Kannan, K., Mabury, S. A., and van Leeuwen, S. P.: Perfluoroalkyl and polyfluoroalkyl substances in the environment: Terminology, classification, and origins, Integr. Environ. Assess. Manag., 7, 513–541, https://doi.org/10.1002/ieam.258, 2011.
Bullard, J. E., Baddock, M., Bradwell, T., Crusius, J., Darlington, E., Gaiero, D., Gassó, S., Gisladottir, G., Hodgkins, R., McCulloch, R., McKenna-Neuman, C., Mockford, T., Stewart, H., and Thorsteinsson, T.: High-latitude dust in the Earth system, Rev. Geophys., 54, 447–485, https://doi.org/10.1002/2016RG000518, 2016.
Busch, J., Ahrens, L., Xie, Z., Sturm, R., and Ebinghaus, R.: Polyfluoroalkyl compounds in the East Greenland Arctic Ocean, J. Environ. Monit., 12, 1242, https://doi.org/10.1039/c002242j, 2010.
Butt, C. M., Muir, D. C. G., Stirling, I., Kwan, M., and Mabury, S. A.: Rapid response of Arctic ringed seals to changes in perfluoroalkyl production, Environ. Sci. Technol., 41, 42–49, https://doi.org/10.1021/es061267m, 2007.
Butt, C. M., Berger, U., Bossi, R., and Tomy, G. T.: Levels and trends of poly- and perfluorinated compounds in the arctic environment, Sci. Total Environ., 408, 2936–2965, https://doi.org/10.1016/j.scitotenv.2010.03.015, 2010.
Cabrerizo, A., Silva, A. De, Muir, D., Spencer, C., Lamoureux, S., and Lafreniere, M.: Transport and Bioaccumulation of Perfluorinated Compounds in an Arctic Lake Catchment Influenced by Permafrost Disturbance and Climate Change, in: SETAC North America 37th Annual Meeting, 2016.
Cai, M., Zhao, Z., Yin, Z., Ahrens, L., Huang, P., Cai, M., Yang, H., He, J., Sturm, R., Ebinghaus, R., and Xie, Z.: Occurrence of Perfluoroalkyl Compounds in Surface Waters from the North Pacific to the Arctic Ocean, Environ. Sci. Technol., 46, 661–668, https://doi.org/10.1021/es2026278, 2012a.
Calafat, A. M., Kuklenyik, Z., Reidy, J. A., Caudill, S. P., Tully, J. S., and Needham, L. L.: Serum Concentrations of 11 Polyfluoroalkyl Compounds in the U.S. Population: Data from the National Health and Nutrition Examination Survey (NHANES) 1999–2000, Environ. Sci. Technol., 41, 2237–2242, https://doi.org/10.1021/es062686m, 2007.
Calvert, J. G., Derwent, R. G., Orlando, J. J., Tyndall, G. S., and Wallington, T. J.: Mechanisms of Atmospheric Oxidation of the Alkanes, Oxford University Press, England,, 2008.
Casal, P., Zhang, Y., Martin, J. W., Pizarro, M., Jiménez, B., and Dachs, J.: Role of Snow Deposition of Perfluoroalkylated Substances at Coastal Livingston Island (Maritime Antarctica), Environ. Sci. Technol., 51, 8460–8470, https://doi.org/10.1021/acs.est.7b02521, 2017.
Criscitiello, A. S., Marshall, S. J., Evans, M. J., Kinnard, C., Norman, A.-L., and Sharp, M. J.: Marine aerosol source regions to Prince of Wales Icefield, Ellesmere Island, and influence from the tropical Pacific, 1979–2001, J. Geophys. Res.-Atmos., 121, 9492–9507, https://doi.org/10.1002/2015JD024457, 2016.
Dassuncao, C., Hu, X. C., Zhang, X., Bossi, R., Dam, M., Mikkelsen, B., and Sunderland, E. M.: Temporal Shifts in Poly- and Perfluoroalkyl Substances (PFASs) in North Atlantic Pilot Whales Indicate Large Contribution of Atmospheric Precursors, Environ. Sci. Technol., 51, 4512–4521, https://doi.org/10.1021/acs.est.7b00293, 2017.
D'eon, J. C., Hurley, M. D., Wallington, T. J., and Mabury, S. A.: Atmospheric chemistry of N-methyl perfluorobutane sulfonamidoethanol, C4F9SO2N(CH3)CH2CH2OH: Kinetics and mechanism of reaction with OH, Environ. Sci. Technol., 40, 1862–1868, https://doi.org/10.1021/es0520767, 2006.
Dinglasan-Panlilio, M. J. A. and Mabury, S. A.: Significant residual fluorinated alcohols present in various fluorinated materials, Environ. Sci. Technol., 40, 1447–1453, https://doi.org/10.1021/es051619+, 2006.
Eichler, A., Schwikowski, M., and Gaggeler, H. W.: Meltwater-induced relocation of chemical species in Alpine firn, Tellus B, 53, 192–203, https://doi.org/10.1034/j.1600-0889.2001.d01-15.x, 2001.
Ellis, D. A., Martin, J. W., De Silva, A. O., Mabury, S. A., Hurley, M. D., Sulbaek Andersen, M. P., and Wallington, T. J.: Degradation of fluorotelomer alcohols: A likely atmospheric source of perfluorinated carboxylic acids, Environ. Sci. Technol., 38, 3316–3321, https://doi.org/10.1021/es049860w, 2004.
Environment and Climate Change Canada: Environmental Performance Agreement, available at: http://ec.gc.ca/epe-epa/default.asp?lang=En&n=AE06B51E-1 (last access: 27 October 2017), 2006.
Eetoolook, J., Koihok, M., Analok, F., Mala, C., Pigalak, T., Igutsaq, D., Angutinguniq, J., Pijamini, A., Aqpik, S., Tattuinee, J., Arnaqjuaq, B., Ivalu, A., Kaunak, J., Tasigat, N., Utok, T., Attungalak, N., Novolinga, Z., Arragutainaq, J. and Kilukishuk, G., Report of the Elder's Conference on Climate Change: 29–31 March 2001, Cambridge Bay, Nunavut, 2001, (Nunavut Tunngavik Incorporated), 2001.
Gascon, G., Sharp, M., Burgess, D., Bezeau, P., and Bush, A. B. G.: Changes in accumulation-area firn stratigraphy and meltwater flow during a period of climate warming: Devon Ice Cap, Nunavut, Canada, J. Geophys. Res.-Earth, 118, 2380–2391, https://doi.org/10.1002/2013JF002838, 2013.
Gewurtz, S. B., Martin, P. A., Letcher, R. J., Burgess, N. M., Champoux, L., Elliott, J. E., and Weseloh, D. V. C.: Spatio-temporal trends and monitoring design of perfluoroalkyl acids in the eggs of gull (Larid) species from across Canada and parts of the United States, Sci. Total Environ., 565, 440–450, https://doi.org/10.1016/j.scitotenv.2016.04.149, 2016.
Groot Zwaaftink, C. D., Grythe, H., Skov, H., and Stohl, A.: Substantial contribution of northern high-latitude sources to mineral dust in the Arctic, J. Geophys. Res.-Atmos., 121, 13678–13697, https://doi.org/10.1002/2016JD025482, 2016.
Heydebreck, F., Tang, J., Xie, Z., and Ebinghaus, R.: Emissions of Per- and Polyfluoroalkyl Substances in a Textile Manufacturing Plant in China and Their Relevance for Workers' Exposure, Environ. Sci. Technol., 50, 10386–10396, https://doi.org/10.1021/acs.est.6b03213, 2016.
Houde, M., Martin, J. W., Letcher, R. J., Solomon, K. R., and Muir, D. C. G.: Biological monitoring of polyfluoroalkyl substances: A review, Environ. Sci. Technol., 40, 3463–3473, https://doi.org/10.1021/es052580b, 2006.
Hung, H., Wong, F., Shoeib, M., Yu, Y., Harner, T., Steffen, A., Muir, D., Teixeira, C., Jantunen, L., Sverko, E., Barresi, E., Fellin, P., Li, H., Green, C., Roach, P., and Wania, F.: Synopsis of Research Conducted Under the 2015–2016 Northern Contaminants Program, available at: http://pubs.aina.ucalgary.ca/ncp/Synopsis20152016SupportingData.pdf (last access: 27 October 2017), 2016.
Jiang, W., Zhang, Y., Yang, L., Chu, X., and Zhu, L.: Perfluoroalkyl acids (PFAAs) with isomer analysis in the commerical PFOS and PFOA products in China, Chemosphere, 127, 180–187, https://doi.org/10.1016/j.chemosphere.2015.01.049, 2015.
Kahl, J. D. W., Martinez, D. A., Kuhns, H., Davidson, C. I., Jaffrezo, J.-L., and Harris, J. M.: Air mass trajectories to Summit, Greenland: A 44-year climatology and some episodic events, J. Geophys. Res. Ocean., 102, 26861–26875, https://doi.org/10.1029/97JC00296, 1997.
Keene, W. C., Pszenny, A. A. P., Galloway, J. N., and Hawley, M. E.: Sea-salt corrections and interpretation of constituent ratios in marine precipitation, J. Geophys. Res., 91, 6647–6658, https://doi.org/10.1029/JD091iD06p06647, 1986.
Kirchgeorg, T., Dreyer, A., Gabrieli, J., Kehrwald, N., Sigl, M., Schwikowski, M., Boutron, C., Gambaro, A., Barbante, C., and Ebinghaus, R.: Temporal variations of perfluoroalkyl substances and polybrominated diphenyl ethers in alpine snow, Environ. Pollut., 178, 367–374, https://doi.org/10.1016/j.envpol.2013.03.043, 2013.
Koerner, R. M.: Mass balance of glaciers in the Queen Elizabeth Islands, Nunavut, Canada, Ann. Glaciol., 42, 417–423, https://doi.org/10.3189/172756405781813122, 2005.
Kwok, K. Y., Yamazaki, E., Yamashita, N., Taniyasu, S., Murphy, M. B., Horii, Y., Petrick, G., Kallenborn, R., Kannan, K., Murano, K., and Lam, P. K. S.: Transport of Perfluoroalkyl substances (PFAS) from an arctic glacier to downstream locations: Implications for sources, Sci. Total Environ., 447, 46–55, https://doi.org/10.1016/j.scitotenv.2012.10.091, 2013.
Lam, J. C. W., Lyu, J., Kwok, K. Y., and Lam, P. K. S.: Perfluoroalkyl Substances (PFASs) in Marine Mammals from the South China Sea and Their Temporal Changes 2002–2014: Concern for Alternatives of PFOS?, Environ. Sci. Technol., 50, 6728–6736, https://doi.org/10.1021/acs.est.5b06076, 2016.
Land, M., de Wit, C. A., Cousins, I. T., Herzke, D., Johansson, J., and Martin, J. W.: What is the effect of phasing out long-chain per- and polyfluoroalkyl substances on the concentrations of perfluoroalkyl acids and their precursors in the environment? A systematic review protocol, Environ. Evid., 4, 1–13, https://doi.org/10.1186/2047-2382-4-3, 2015.
Legrand, M. and Mayewski, P.: Glaciochemistry of polar ice cores: A review, Rev. Geophys., 35, 219–243, https://doi.org/10.1029/96RG03527, 1997.
Lim, T. C., Wang, B., Huang, J., Deng, S., and Yu, G.: Emission Inventory for PFOS in China: Review of Past Methodologies and Suggestions, Sci. World J., 11, 1963–1980, https://doi.org/10.1100/2011/868156, 2011.
Luo, C., Mahowald, N. M., and del Corral, J.: Sensitivity study of meteorological parameters on mineral aerosol mobilization, transport, and distribution, J. Geophys. Res., 108, 4447, https://doi.org/10.1029/2003JD003483, 2003.
MacInnis, J. J., French, K., Muir, D. C. G., Spencer, C., Criscitiello, A., De Silva, A. O., and Young, C. J.: A 14-year depositional ice record of perfluoroalkyl substances in the High Arctic, Environ. Sci. Process. Impacts, 19, 22–30, https://doi.org/10.1039/C6EM00593D, 2017.
Martin, J. W., Ellis, D. A., Mabury, S. A., Hurley, M. D., and Wallington, T. J.: Atmospheric Chemistry of Perfluoroalkanesulfonamides: Kinetic and Product Studies of the OH Radical and Cl Atom Initiated Oxidation of N-Ethyl Perfluorobutanesulfonamide, Environ. Sci. Technol., 40, 864–872, https://doi.org/10.1021/es051362f, 2006.
McMurdo, C. J., Ellis, D. A., Webster, E., Butler, J., Christensen, R. D., and Reid, L. K.: Aerosol Enrichment of the Surfactant PFO and Mediation of the Water-Air Transport of Gaseous PFOA, Environ. Sci. Technol., 42, 3969–3974, https://doi.org/10.1021/es7032026, 2008.
Meyer, T., Muir, D. C. G., Teixeira, C., Wang, X., Young, T., and Wania, F.: Deposition of Brominated Flame Retardants to the Devon Ice Cap, Nunavut, Canada, Environ. Sci. Technol., 46, 826–833, https://doi.org/10.1021/es202900u, 2012.
Miller, J. E., Kahl, J. D. W., Heller, F., and Harris, J. M.: A three-dimensional residence-time analysis of potential summertime atmospheric transport to Summit, Greenland, Ann. Glaciol., 35, 403–408, https://doi.org/10.3189/172756402781816663, 2002.
Mochizuki, T., Kawamura, K., Aoki, K., and Sugimoto, N.: Long-range atmospheric transport of volatile monocarboxylic acids with Asian dust over a high mountain snow site, central Japan, Atmos. Chem. Phys., 16, 14621–14633, https://doi.org/10.5194/acp-16-14621-2016, 2016.
NSIDC: Arctic Sea Ice Minimum, National Snow and Ice Data Center, available at: https://climate.nasa.gov/vital-signs/arctic-sea-ice/ (last access: 1 March 2018), 2017.
Olsen, G. W., Church, T. R., Miller, J. P., Burris, J. M., Hansen, K. J., Lundberg, J. K., Armitage, J. B., Herron, R. M., Medhdizadehkashi, Z., Nobiletti, J. B., O'Neill, E. M., Mandel, J. H., and Zobel, L. R.: Perfluorooctanesulfonate and Other Fluorochemicals in the Serum of American Red Cross Adult Blood Donors, Environ. Health Persp., 111, 1892–1901, https://doi.org/10.1289/ehp.6316, 2003.
Paul, A. G., Jones, K. C., and Sweetman, A. J.: A First Global Production, Emission, And Environmental Inventory For Perfluorooctane Sulfonate, Environ. Sci. Technol., 43, 386–392, https://doi.org/10.1021/es802216n, 2009.
Pinglot, J. F., Vaikmäe, R. A., Kamiyama, K., Igarashi, M., Fritzsche, D., Wilhelms, F., Koerner, R., Henderson, L., Isaksson, E., Winther, J.-G., Van de wal, R. S. W., Fournier, M., Bouisset, P., and Meijer, H. A. J.: Ice cores from Arctic sub-polar glaciers: chronology and post-depositional processes deduced from radioactivity measurements, J. Glaciol., 49, 149–158, https://doi.org/10.3189/172756503781830944, 2003.
Plassmann, M. M., Meyer, T., Lei, Y. D., Wania, F., McLachlan, M. S., and Berger, U.: Laboratory Studies on the Fate of Perfluoroalkyl Carboxylates and Sulfonates during Snowmelt, Environ. Sci. Technol., 45, 6872–6878, https://doi.org/10.1021/es201249d, 2011.
Preunkert, S., Legrand, M., and Wagenbach, D.: Causes of enhanced fluoride levels in Alpine ice cores over the last 75 years: Implications for the atmospheric fluoride budget, J. Geophys. Res.-Atmos., 106, 12619–12632, https://doi.org/10.1029/2000JD900755, 2001.
Prevedouros, K., Cousins, I. T., Buck, R. C., and Korzeniowski, S. H.: Sources, Fate and Transport of Perfluorocarboxylates, Environ. Sci. Technol., 40, 32–44, https://doi.org/10.1021/es0512475, 2006.
Rahn, K. A., Borys, R. D., and Shaw, G. E.: The Asian source of Arctic haze bands, Nature, 268, 713–715, https://doi.org/10.1038/268713a0, 1977.
Readinger, C.: Ice Core Proxy Methods for Tracking Climate Change, CSA Discov. Guid., 1–10 available at: http://www.csa.com/discoveryguides/discoveryguides-main.php (last access: 27 October 2017), 2006.
Reth, M., Berger, U., Broman, D., Cousins, I. T., Nilsson, E. D., and McLachlan, M. S.: Water-to-air transfer of perfluorinated carboxylates and sulfonates in a sea spray simulator, Environ. Chem., 8, 381–388, https://doi.org/10.1071/EN11007, 2011.
Ruisheng, Y.: Preliminary Information on Risk Management Evaluation of PFOS's in China, Ministry of Environmental Protection of China, 2008.
Schenker, U., Scheringer, M., MacLeod, M., Martin, J. W., Cousins, I. T., and Hungerbühler, K.: Contribution of Volatile Precursor Substances to the Flux of Perfluorooctanoate to the Arctic, Environ. Sci. Technol., 42, 3710–3716, https://doi.org/10.1021/es703165m, 2008.
Scheringer, M., Trier, X., Cousins, I. T., de Voogt, P., Fletcher, T., Wang, Z., and Webster, T. F.: Helsingør Statement on poly- and perfluorinated alkyl substances (PFASs), Chemosphere, 114, 337–339, https://doi.org/10.1016/j.chemosphere.2014.05.044, 2014.
Sharp, M., Burgess, D. O., Cogley, J. G., Ecclestone, M., Labine, C., and Wolken, G. J.: Extreme melt on Canada's Arctic ice caps in the 21st century, Geophys. Res. Lett., 38, L11501, https://doi.org/10.1029/2011GL047381, 2011.
Shoeib, M., Harner, T., and Vlahos, P.: Perfluorinated Chemicals in the Arctic Atmosphere, Environ. Sci. Technol., 40, 7577–7583, https://doi.org/10.1021/es0618999, 2006.
Stahl, T., Mattern, D., and Brunn, H.: Toxicology of perfluorinated compounds, Environ. Sci. Eur., 23, 38, https://doi.org/10.1186/2190-4715-23-38, 2011.
Stein, A. F., Draxler, R. R., Rolph, G. D., Stunder, B. J. B., Cohen, M. D., and Ngan, F.: NOAA's HYSPLIT Atmospheric Transport and Dispersion Modeling System, B. Am. Meteorol. Soc., 96, 2059–2077, https://doi.org/10.1175/BAMS-D-14-00110.1, 2015.
Steinlin, C., Bogdal, C., Lüthi, M. P., Pavlova, P. A., Schwikowski, M., Zennegg, M., Schmid, P., Scheringer, M., and Hungerbühler, K.: A Temperate Alpine Glacier as a Reservoir of Polychlorinated Biphenyls: Model Results of Incorporation, Transport, and Release, Environ. Sci. Technol., 50, 5572–5579, https://doi.org/10.1021/acs.est.5b05886, 2016.
Stock, N. L., Furdui, V. I., Muir, D. C. G., and Mabury, S. A.: Perfluoroalkyl contaminants in the Canadian Arctic: Evidence of atmospheric transport and local contamination, Environ. Sci. Technol., 41, 3529–3536, https://doi.org/10.1021/es062709x, 2007.
Sullivan, R. C., Guazzotti, S. A., Sodeman, D. A., and Prather, K. A.: Direct observations of the atmospheric processing of Asian mineral dust, Atmos. Chem. Phys., 7, 1213–1236, https://doi.org/10.5194/acp-7-1213-2007, 2007.
Thackray, C. P. and Selin, N. E.: Uncertainty and variability in atmospheric formation of PFCAs from fluorotelomer precursors, Atmos. Chem. Phys., 17, 4585–4597, https://doi.org/10.5194/acp-17-4585-2017, 2017.
US EPA: PFOA Stewardship Program Baseline Year Summary Report, available at: https://www.epa.gov/assessing-and-managing-chemicals-under-tsca/epas-non-cbi-summary-tables-2015-company-progress (last access: 6 April 2018), 2016.
Wallington, T. J., Hurley, M. D., Xia, J., Wuebbles, D. J., Sillman, S., Ito, A., Penner, J. E., Ellis, D. A., Martin, J., Mabury, S. A., Nielsen, O. J., and Sulbaek Andersen, M. P.: Formation of C7F15COOH (PFOA) and Other Perfluorocarboxylic Acids during the Atmospheric Oxidation of 8:2 Fluorotelomer Alcohol, Environ. Sci. Technol., 40, 924–930, https://doi.org/10.1021/es051858x, 2006.
Wang, T., Vestergren, R., Herzke, D., Yu, J. ,and Cousins, I. T.: Levels, Isomer Profiles, and Estimated Riverine Mass Discharges of Perfluoroalkyl Acids and Fluorinated Alternatives at the Mouths of Chinese Rivers, Environ. Sci. Technol., 50, 11584–11592, https://doi.org/10.1021/acs.est.6b03752, 2016.
Wang, X., Halsall, C., Codling, G., Xie, Z., Xu, B., Zhao, Z., Xue, Y., Ebinghaus, R., and Jones, K. C.: Accumulation of Perfluoroalkyl Compounds in Tibetan Mountain Snow: Temporal Patterns from 1980 to 2010, Environ. Sci. Technol., 48, 173–181, https://doi.org/10.1021/es4044775, 2014.
Wang, Z., Cousins, I. T., Scheringer, M., and Hungerbühler, K.: Fluorinated alternatives to long-chain perfluoroalkyl carboxylic acids (PFCAs), perfluoroalkane sulfonic acids (PFSAs) and their potential precursors, Environ. Int., 60, 242–248, https://doi.org/10.1016/j.envint.2013.08.021, 2013.
Wang, Z., Cousins, I. T., Scheringer, M., Buck, R. C., and Hungerbuhler, K.: Global emission inventories for C4-C14 perfluoroalkyl carboxylic acid (PFCA) homologues from 1951 to 2030, Part 1: Production and emissions from quantifiable sources, Environ. Int., 70, 62–75, https://doi.org/10.1016/j.envint.2014.04.013, 2014a.
Wang, Z., Cousins, I. T., Scheringer, M., Buck, R. C., and Hungerbühler, K.: Global emission inventories for C4–C14 perfluoroalkyl carboxylic acid (PFCA) homologues from 1951 to 2030, part II: The remaining pieces of the puzzle, Environ. Int., 69, 166–176, https://doi.org/10.1016/j.envint.2014.04.006, 2014b.
Wang, Z., Boucher, J. M., Scheringer, M., Cousins, I. T., and Hungerbühler, K.: Toward a Comprehensive Global Emission Inventory of C4–C10 Perfluoroalkanesulfonic Acids (PFSAs) and Related Precursors: Focus on the Life Cycle of C8-Based Products and Ongoing Industrial Transition, Environ. Sci. Technol., 51, 4482–4493, https://doi.org/10.1021/acs.est.6b06191, 2017.
Weppner, W. A.: 3M Company, Phase-out plan for POSF-based products; US Environmental Protection Agency, 2000.
Xie, S., Wang, T., Liu, S., Jones, K. C., Sweetman, A. J., and Lu, Y.: Industrial source identification and emission estimation of perfluorooctane sulfonate in China, Environ. Int., 52, 1–8, https://doi.org/10.1016/j.envint.2012.11.004, 2013.
Xu, Z., Li, L., Henkelmann, B., and Schramm, K.-W.: Occurrence of fluorotelomer alcohols at two Alpine summits: sources, transport and temporal trends, Environ. Chem., 14, 215–223, https://doi.org/10.1071/EN16190, 2017.
Yao, Y., Sun, H., Gan, Z., Hu, H., Zhao, Y., Chang, S., and Zhou, Q.-X.: Nationwide Distribution of Per- and Polyfluoroalkyl Substances in Outdoor Dust in Mainland China From Eastern to Western Areas, Environ. Sci. Technol., 50, 3676–3685, https://doi.org/10.1021/acs.est.6b00649, 2016.
Yarwood, G., Kemball-Cook, S., Keinath, M., Waterland, R. L., Korzeniowski, S. H., Buck, R. C., Russell, M. H., and Washburn, S. T.: High-Resolution Atmospheric Modeling of Fluorotelomer Alcohols and Perfluorocarboxylic Acids in the North American Troposphere, Environ. Sci. Technol., 41, 5756–5762, https://doi.org/10.1021/es0708971, 2007.
Young, C. J. and Mabury, S. A.: Atmospheric Perfluorinated Acid Precursors: Chemistry, Occurrence, and Impacts, edited by: Whitacre, D. M., Springer New York, New York, NY, 2010.
Young, C. J., Furdui, V. I., Franklin, J., Koerner, R. M., Muir, D. C. G., and Mabury, S. A.: Perfluorinated acids in Arctic snow: New evidence for atmospheric formation, Environ. Sci. Technol., 41, 3455–3461, https://doi.org/10.1021/es0626234, 2007.
Zdanowicz, C. M., Zielinski, G. A., and Wake, C. P.: Characteristics of modern atmospheric dust deposition in snow on the Penny Ice Cap, Baffin Island, Arctic Canada, Tellus B, 50, 506–520, https://doi.org/10.1034/j.1600-0889.1998.t01-1-00008.x, 1998.
Zhao, Z., Xie, Z., Möller, A., Sturm, R., Tang, J., Zhang, G., and Ebinghaus, R.: Distribution and long-range transport of polyfluoroalkyl substances in the Arctic, Atlantic Ocean and Antarctic coast, Environ. Pollut., 170, 71–77, https://doi.org/10.1016/j.envpol.2012.06.004, 2012.