the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 13 Mar 2018
| 13 Mar 2018
Ubiquitous influence of wildfire emissions and secondary organic aerosol on summertime atmospheric aerosol in the forested Great Lakes region
Matthew J. Gunsch
Nathaniel W. May
Miao Wen
Courtney L. H. Bottenus
Daniel J. Gardner
Timothy M. VanReken
Steven B. Bertman
Philip K. Hopke
Long-range aerosol transport affects locations hundreds of kilometers from the point of emission, leading to distant particle sources influencing rural environments that have few major local sources. Source apportionment was conducted using real-time aerosol chemistry measurements made in July 2014 at the forested University of Michigan Biological Station near Pellston, Michigan, a site representative of the remote forested Great Lakes region. Size-resolved chemical composition of individual 0.5–2.0 µm particles was measured using an aerosol time-of-flight mass spectrometer (ATOFMS), and non-refractory aerosol mass less than 1 µm (PM1) was measured with a high-resolution aerosol mass spectrometer (HR-AMS). The field site was influenced by air masses transporting Canadian wildfire emissions and urban pollution from Milwaukee and Chicago. During wildfire-influenced periods, 0.5–2.0 µm particles were primarily aged biomass burning particles (88 % by number). These particles were heavily coated with secondary organic aerosol (SOA) formed during transport, with organics (average O∕C ratio of 0.8) contributing 89 % of the PM1 mass. During urban-influenced periods, organic carbon, elemental carbon–organic carbon, and aged biomass burning particles were identified, with inorganic secondary species (ammonium, sulfate, and nitrate) contributing 41 % of the PM1 mass, indicative of atmospheric processing. With current models underpredicting organic carbon in this region and biomass burning being the largest combustion contributor to SOA by mass, these results highlight the importance for regional chemical transport models to accurately predict the impact of long-range transported particles on air quality in the upper Midwest, United States, particularly considering increasing intensity and frequency of Canadian wildfires.
- Article
(6218 KB) -
Supplement
(776 KB) - BibTeX
- EndNote





Atmospheric particulate matter less than 2.5 µm in diameter (PM2.5) has significant impacts on air quality, climate, and human health (Calvo et al., 2013; Pöschl and Shiraiwa, 2015). Atmospheric particles directly affect climate by scattering incoming solar radiation and indirectly by acting as cloud condensation nuclei (CCN) and ice nuclei (IN) (IPCC, 2013). Increased levels of PM2.5 are also linked to increased health risks, particularly respiratory and cardiovascular diseases (Brook et al., 2004; Pope and Dockery, 2006). Particles can impact areas hundreds of kilometers from their sources through long-range transport, with residence times of up to 2 weeks depending on particle size and chemical composition (Uno et al., 2009). Determining the impact of the long-range transported particles, as well as how they are transformed in the atmosphere during transport, is a critical topic to accurately predict their air quality and climate effects (Ault et al., 2011; Creamean et al., 2013). During transport, particles undergo heterogeneous reactions and gas–particle partitioning, aging the particles and leading to primary particles (e.g., soot) becoming internally mixed with secondary species, including water, ammonium, nitrate, sulfate, and oxidized organic carbon (OC), thus changing the chemical composition of individual particles (Moffet and Prather, 2009; Riemer and West, 2013). These aging processes are particularly important since chemical composition is directly related to particle properties, including reactivity, hygroscopicity, toxicity, scattering, and absorption properties (Brook et al., 2004; Pöschl, 2005; Calvo et al., 2013; Fierce et al., 2016). Particle properties also differ based on the distribution of chemical species, or mixing state, within a population of particles – whether various chemical species are contained within a single particle (internally mixed) or within different particles (externally mixed). Particle mixing state representation in models is particularly important (Bauer et al., 2013), especially for predicting aerosol impacts on the climate (Matsui et al., 2013; Fierce et al., 2016).
Long-range transport of atmospheric particles can contribute to both remote and populated locations being out of compliance with air quality regulations (National Research Council and National Academies, 2010). For example, elevated aerosol mass and ozone in Europe, eastern Canada, and the northeastern United States has been attributed to transported Canadian wildfire emissions (Forster et al., 2001; Colarco et al., 2004; Müller et al., 2005; Y. Wang et al., 2010b; Dutkiewicz et al., 2011; Miller et al., 2011; Dempsey, 2013; Kang et al., 2014; Dreessen et al., 2016). A multiday exceedance of the National Ambient Air Quality Standard for ozone in Maryland during the summer of 2015 was attributed to Canadian wildfire emissions (Dreessen et al., 2016). Similarly, elevated PM2.5 observed in New York and Wisconsin has been attributed to Ohio River valley emissions. Transported pollutants can also impact biogenic secondary organic aerosol (SOA) formation in remote locations (Carlton et al., 2010; Emanuelsson et al., 2013; Xu et al., 2015; Rattanavaraha et al., 2016). Finally, prior and ongoing studies through the IMPROVE program in rural locations throughout North America have investigated both transported and local contributions to the aerosol populations (Hand et al., 2011). Uncertainty in the contributions of long-range aerosols and limited measurements in remote areas can lead to inaccuracies in modeling of aerosol source contributions.
Relatively few studies have chemically characterized atmospheric aerosols in the rural Great Lakes region of the United States (Sheesley et al., 2004; Kim et al., 2005, 2007; Zhang et al., 2009; Jeong et al., 2011; Sjostedt et al., 2011; Kundu and Stone, 2014; Bullard et al., 2017). Except for the major metropolitan areas of Detroit (MI), Chicago (IL), Minneapolis (MN), and Milwaukee (WI), much of the land is characterized by rural agricultural areas and remote forests without significant anthropogenic emissions. A study in the upper peninsula of Michigan conducted by Sheesley et al. (2004) observed major contributions from SOA from both biogenic and anthropogenic volatile organic compound (VOC) oxidation in the summer. Studies across rural Illinois and Ohio found major atmospheric contributions from secondary sulfate, nitrate, and OC, consistent with aerosol aging during transport (Kim et al., 2005, 2007; Zhang et al., 2009), though these locations were much less forested than the more northern Great Lakes regions. Kundu and Stone (2014) measured composition and sources at rural locations in Iowa, identifying major PM mass contributions from biomass burning, combustion, and dust. Jeong et al. (2011), Sjostedt et al. (2011), and Slowik et al. (2011) identified contributions from SOA, elemental carbon, and dust in rural Harrow, Ontario, downwind of Detroit and Windsor. The scarcity of measurement data in the rural Great Lakes region provides limited opportunities for model evaluation and requires assumptions of background primary aerosol.
In remote regions, there are challenges in distinguishing and identifying primary and secondary aerosol sources, particularly for bulk methods (Pratt and Prather, 2012). Single-particle mass spectrometry allows the identification of particle sources through comparisons with source “fingerprints” and particle aging through characterization of individual particle chemical mixing state (Pratt and Prather, 2009, 2012). Therefore, to apportion the sources of the aerosol population influencing remote northern Michigan, single-particle mass spectrometry measurements were conducted during July 2014 at the University of Michigan Biological Station (UMBS) near Pellston, MI. In this study, individual particle chemical composition, measured in real time using single-particle mass spectrometry, was used to identify the sources and secondary processing of transported particles at UMBS. In addition, high-resolution aerosol mass spectrometry (HR-AMS) measured chemically resolved mass concentrations of non-refractory aerosol (organics, sulfate, nitrate, ammonium, and chloride) to provide complementary mass-based characterization of the transported particles at UMBS.
2.1 Field site and instrumentation
Atmospheric measurements were conducted from 13 to 24 July 2014 at UMBS near Pellston, MI, a 10 000-acre (4050 ha) remote, forested location with little local pollution (Carroll et al., 2001). The closest major cities are Milwaukee (370 km southwest), Detroit (385 km south), and Chicago (466 km southwest). Instrumentation was located within a laboratory at the base of the Program for Research on Oxidants: Photochemistry, Emissions, and Transport (PROPHET) tower, a 30 m tall sampling tower (45∘33′31 N, 84∘42′52 W) (Carroll et al., 2001). Air was sampled from 34 (∼14 m above the forest canopy) through foam-insulated 1.09 cm I.D. copper tubing at a flow rate of 9.25 L min−1 (laminar) with a residence time of 15 s. This tubing was connected to a shared sampling manifold at the base of the tower, allowing individual instruments to each have a dedicated sampling line while limiting particle loss.
An aerosol time-of-flight mass spectrometer (ATOFMS, model 3800, TSI, Inc., Shoreview, MN) (Gard et al., 1997; Dall'Osto et al., 2004), described briefly below, was used to measure the size and chemical composition of individual atmospheric particles ranging from 0.5 to 2.0 µm in vacuum aerodynamic diameter (dva) (Sect. 2.2). An Aerodyne HR-AMS (DeCarlo et al., 2006) measured chemically resolved mass concentrations of non-refractory fine-particulate material (nominal vacuum aerodynamic diameter range of 0.05–1.0 µm) from 15 to 24 July 2014. Concentrations for major composition classes (organics, sulfate, nitrate, ammonium, and chloride) are reported here. O∕C ratios were calculated throughout the study using the methods described by Canagaratna et al. (2015). The operation of the HR-AMS followed standard practice as described elsewhere (Jayne et al., 2000; Allan et al., 2003; Jimenez et al., 2003; Allan et al., 2004); the sampling resolution for the UMBS observations was 2.5 min. Calibrations for instrument flow rate, particle sizing and transmission, and ionization efficiency were conducted during the study following documented procedures (Jimenez and DeCarlo, 2017). Data were analyzed using SQUIRREL (version 1.60) and the high resolution analysis software tool PIKA (version 1.20) (Sueper, 2010), with the concentrations corrected based on the estimated composition-dependent collection efficiency (Middlebrook et al., 2012). Additional instrumentation included an ozone analyzer (Thermo Scientific model 49), a scanning mobility particle sizer spectrometer (SMPS, TSI model 3936) with a sheath flow rate of 4 L min−1 and an aerosol flow rate of 0.4 L min−1 for measuring size-resolved number concentrations of mobility diameter particles 12–600 nm, and an aerodynamic particle sizer spectrometer (APS, TSI model 3321) for measuring size-resolved number concentrations of 0.5–19 µm aerodynamic diameter particles. SMPS and APS size distributions were merged to give a continuous aerosol distribution from 0.01 to 2.5 µm (aerodynamic diameter) using previously established methods (Khlystov et al., 2004), assuming a density of 1.5 g cm−3 and shape factor of 1.
2.2 Aerosol time-of-flight mass spectrometer (ATOFMS)
Using the ATOFMS, 11 430 individual atmospheric particles ranging from 0.5 to 2.0 µm in dva were chemically analyzed from 13 to 24 July 2014. The design and operation of the ATOFMS has been described in detail elsewhere (Dall'Osto et al., 2004; Su et al., 2004). Briefly, particles are focused through an aerodynamic lens system and optically detected by two 532 nm continuous wave lasers spaced 6 cm apart. Particle aerodynamic diameter is determined from particle velocity, which was calibrated using monodispersed spherical polystyrene latex spheres (0.4–2.5 µm, Polysciences, Inc.) of known diameter and density. Particles are individually desorbed and ionized by a 266 nm Nd:YAG laser that was operated at ∼1.2 mJ, and the resulting ions enter a dual-polarity reflectron time-of-flight mass spectrometer. Positive and negative ion mass spectra corresponding to the same individual particles are collected. Mass spectral peak lists for individual particles were generated using TSI MS-Analyze software.
The individual particle mass spectra were analyzed using YAADA (yaada.org), a software toolkit for MATLAB. Particles were clustered in YAADA using the ART-2a algorithm with a vigilance factor of 0.80 and a learning rate of 0.05 for 20 iterations (Song et al., 1999). The top 50 clusters were manually classified into five particle types described in Sect. 3.1. These top 50 clusters contained 92 % of the 11 430 particle mass spectra collected and are the focus of the paper. Particle identification was based on characteristic ATOFMS mass spectral signatures previously described (Silva et al., 1999; Pastor et al., 2003; Qin et al., 2012). The errors associated with number fractions for each particle type were calculated using binomial statistics.
To obtain chemically resolved number and mass concentrations for 0.5–2.0 µm particles, ATOFMS particle counts were scaled with the APS size-resolved particle number concentration data using the method of Qin et al. (2006) to account for size-dependent particle transmission in the inlet. Briefly, ratios of APS number concentration to ATOFMS non-scaled number concentration were calculated every 3 h for each individual size bin defined by the APS for use as scaling factors. Each scaling factor was then multiplied by the corresponding ATOFMS number concentration, providing size and chemically resolved particle number concentrations for each of the four particle types. These number concentrations were then converted to mass concentrations using assumed spherical shape and compositionally specific densities. The following densities were applied for the four particle types: 1.5 g cm−3 for biomass burning, 1.5 g cm−3 for salts, and 1.25 g cm−3 for organic carbon–sulfate (OC-sulfate) and elemental carbon–organic carbon–sulfate (ECOC-sulfate) particles (Spencer et al., 2007; Moffet et al., 2008).
3.1 Overview
The UMBS campaign (13–24 July 2014) was characterized by air masses from three primary directions: north, northwest, and southwest (Figs. S1 and S2 in the Supplement), representative of periods observed during previous UMBS summer studies (Cooper et al., 2001; VanReken et al., 2015). Analysis of NOAA HYSPLIT backward air mass trajectories showed four distinct air mass time periods (Fig. S2). From 13 to 15 July, air primarily came from northwestern Canada. From 15 to 17 July, the wind shifted and came from directly north crossing over Lake Superior and Lake Michigan before arriving at the field site. In contrast, from 17 to 22 July the air came mainly from south-southwest of the field site, crossing over the major metropolitan areas of Chicago and Milwaukee followed by Lake Michigan. Finally, from 23 to 24 July, air came from the north-northwest of the field site, crossing Lake Superior and Lake Michigan from northern Canada (Fig. S2). During summer 2009, VanReken et al. (2015) found that 60 % of the air masses came from the north-northwest of UMBS, similar to this study (57 %). Air came from southern polluted regions 43 % of the time during our study, compared to 29 % during July–August 2009 (VanReken et al., 2015).

Figure 1Time-resolved PM2.5 number and mass concentrations and ozone mole ratios during the different periods of air mass influence. Periods without data are due to instrument downtime. Colors of the different time periods correspond to the colors of the corresponding HYSPLIT backward air mass trajectories in Fig. S2.

Figure 2Average positive and negative ion single-particle mass spectra (ATOFMS), with characteristic peaks labeled, for the dominant aged combustion particle types observed: (a) biomass burning, (b) OC-sulfate, and (c) ECOC-sulfate.

Figure 3PM1 non-refractory chemically speciated mass concentrations, as well as O∕C ratios (20 min averages), measured with the HR-AMS. Periods of influence are notated and separated by solid vertical lines. Pie charts represent the average mass fractions for each air mass period, with average O∕C ratio inset.
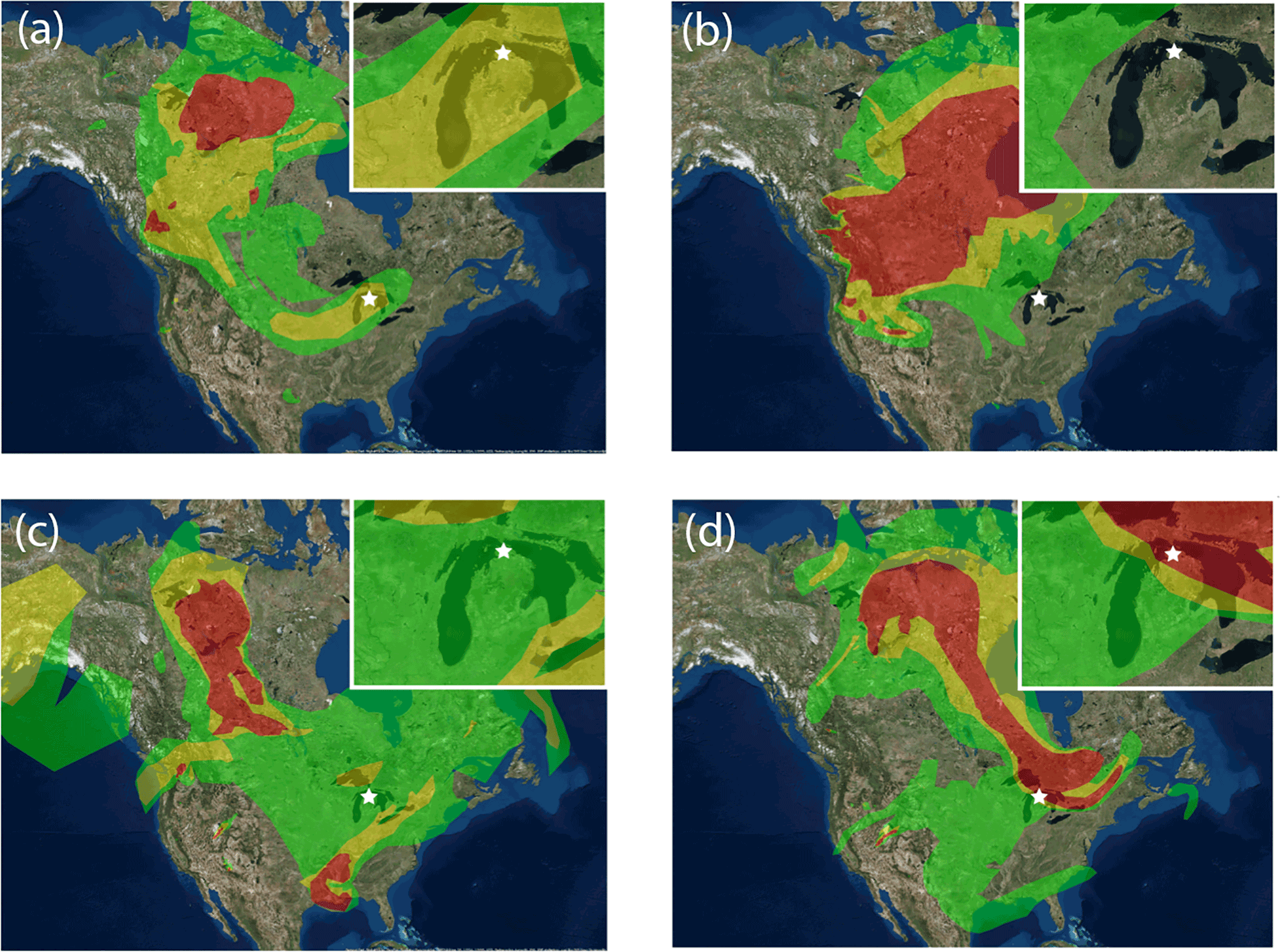
Figure 4Representative NOAA HMS smoke maps for four representative days during the time periods of different air influence: (a) 14 July, wildfire influence; (b) 16 July, remote background influence; (c) 21 July, urban influence; (d) 24 July, wildfire influence. Inset enlarges the state of Michigan to clearly display smoke influence on the field site, shown as a star.
Total PM2.5 number, PM2.5 mass, and ozone concentrations ranged from 143 to 6031 particles cm−3 (average ± SD: 1822±1181 particles cm−3), 1 to 43 µg m−3 (average ± SD: 8±8 µg m−3), and 9 to 63 ppb (average ± SD: 32±14 ppb), respectively (Fig. 1). Maximum concentrations were detected when the air arrived from the southwestern urban areas, and the minimum values were observed for air masses from the north during remote air transport (Figs. S3 and S4). Previously, VanReken et al. (2015) observed an 85 % increase in particle number concentration when air originating from these southwestern urban areas impacted UMBS. These results suggest a wide range of influencing sources, which were observed by the ATOFMS. Here, we examine the influences of wildfires (Sect. 3.3) and urban pollution (Sect. 3.4) on summertime aerosol chemical composition, compared to remote background air mass influence (Sect. 3.2), at UMBS.
Major individual particle types observed by ATOFMS included biomass burning, OC-sulfate, and ECOC-sulfate (Fig. 2). Biomass burning particles were characterized by intense peaks at m∕z 39 (K+) and −97 (), as well as less intense peaks at m∕z 12 (C+), 18 (), and 27 () (Pratt et al., 2010). Biomass burning particles also contained a peak at m∕z 43 (C2H3O+), a marker for oxidized OC, which is addressed further in Sect. 3.3. Biomass burning was the most prominent particle type, comprising ∼80 % of submicron (0.5–1.0 µm) and ∼50 % of supermicron (1–2 µm) particles, by number, throughout the study, with the number fraction varying according to the level of influence from wildfires. OC-sulfate particles contributed ∼7 %, by number, to submicron (0.5–1.0 µm) particles and ∼8 %, by number, to supermicron (1.0–2.0 µm) particles and were characterized by intense peaks at m∕z 27 (), 39 (), ±43 (C2H3O±), and −97 (). OC-sulfate particles can originate from a variety of sources including primary vehicular emissions (Toner et al., 2008) and secondary organic sources (Pratt and Prather, 2009). The intense m∕z 43 (most intense OC-sulfate particle ion peak) is indicative of significant SOA coatings on combustion particles, including biomass burning (Pratt and Prather, 2009). ECOC-sulfate particles, characterized by fragment peaks, observed at m∕z 12 (C+), 24 (), 36 (), 48 (), etc., as well as markers at m∕z 27 (), 18 (), and −97 (), are attributed to vehicular emissions (Toner et al., 2006, 2008) and contributed ∼5 %, by number, to both sub- and supermicron particles, with the majority observed on 22 July during an urban-influenced air mass. In addition to the previously mentioned combustion and secondary particles, Na and Ca salts internally mixed with nitrate were episodically detected, primarily during 16–18 July and 24–25 July. Based on elemental ratios and established mass spectral fingerprints, these salts originated from the Great Lakes (Axson et al., 2016; May et al., 2018) and/or seawater, rather than mineral dust (Sullivan et al., 2007; Ault et al., 2011; Fitzgerald et al., 2015), and are the focus of an upcoming paper. For each of the discussed particle types, we present the chemical mixing state by reporting the number percentage of particles within each particle type that contain a mass spectral marker corresponding to each secondary aerosol chemical species of interest, including sulfate (, m∕z −97), nitrate (, m∕z −46, and/or , m∕z −62), ammonium (, m∕z 18), and oxidized OC (C3H2O−, m∕z −43, or C3H2O+, m∕z 43) (Qin et al., 2012).
PM1 mass measured with the HR-AMS was on average 73 % organics (7.8 µg m−3) throughout the study, with a substantial contribution from oxidized organics as determined by an average HR-AMS O∕C ratio of 0.84 and through the ATOFMS oxidized OC ion marker m∕z 43, C2H3O+ (Aiken et al., 2008; Qin et al., 2012). O∕C ratios between 0.6 and 1 are commonly associated with low-volatility oxidized organic aerosol (LV-OOA) that has undergone extensive aging (Jimenez et al., 2009), consistent with the single-particle observation that SOA coated the major particle types. In addition, the ammonium balance of predicted ammonium vs. measured ammonium throughout the study (Fig. S5) shows a slight deficit in measured ammonium, typically indicative of acidic aerosol or the presence of organic nitrates and/or sulfates (Farmer et al., 2010). Also consistent with atmospheric processing during long-range transport, 92 % of all 0.5–2.0 µm particles, by number, were measured with the ATOFMS to be internally mixed with secondary species, including sulfate (, m∕z −97), nitrate (, m∕z −46 and/or , m∕z −62), ammonium (, m∕z 18), and/or oxidized OC (C3H2O−, m∕z −43 or C3H2O+, m∕z 43) (Qin et al., 2012). On average, sulfate comprised 20 % (2.2 µg m−3) of the total PM1 mass measured with the HR-AMS.
3.2 Remote background air mass influence
From 15 to 17 July, air arrived at UMBS originating from rural northern Canada. This remote background air mass period was differentiated from the wildfire-influenced periods (Sect. 3.3) based on the lack of smoke impacting the site, as indicated by NOAA Smoke and Fire products (Fig. 4). The average PM2.5 number concentration was 903±499 particles cm−3 (range of 143–2163 particles cm−3, Fig. 1), and the average PM2.5 mass concentration was 1.9±0.4 µg m−3 with a particle number mode of 82 nm (Fig. 1 and S3), comparable to the average particle number (1630±1280 cm−3) and mode (73±21 nm) observed by VanReken et al. (2015) during background air mass influence at UMBS in summer 2009. The average ozone concentration was 17±6 ppb (Fig. 1), similar to previous measurements at UMBS during background air mass influence (Cooper et al., 2001). Despite the lack of direct wildfire influence (Fig. 4), 61±1 % of the 0.5–2.0 µm particles, by number, were classified by ATOFMS as aged biomass burning aerosols, relatively similar to the background biomass burning particle influence reported by Hudson et al. (2004) and Pratt et al. (2010) for the United States free troposphere (33–52 %, by number). Biomass burning particles were internally mixed with oxidized OC (80±2 %, by number) and/or mixed with sulfate (85±2 %). Nitrate was internally mixed with 8±2 %, by number, of biomass burning particles and 33±3 %, by number, of OC-sulfate particles. It is likely that, while the observed biomass burning particles have a small potassium-rich (biomass burning) core, they are primarily SOA by mass (Pratt and Prather, 2009; Moffet et al., 2010) (Sect. 3.3). The HR-AMS showed average PM1 organic mass concentrations of 4.4 µg m−3, with minimal contribution from sulfate (0.3 µg m−3), as well as nitrate and ammonium (both less than 0.1 µg m−3 on average) (Fig. 3).
The significant internal mixing of oxidized OC combined with the significant organic mass loading (average HR-AMS O∕C ratio of 0.9) is consistent with high SOA mass on the particles (Aiken et al., 2008). Previous studies in rural and forested environments found similarly high O∕C ratios during periods of unpolluted air and attributed this to regional SOA formation (Jimenez et al., 2009; Sun et al., 2009; Raatikainen et al., 2010; Sjostedt et al., 2011). There was a notable spike in O∕C ratio on 15–16 July to 1.2, indicative of very highly oxidized organics. O∕C ratios of this magnitude have previously been observed at the remote Whistler Mountain, where organic aerosol O∕C ratios up to ∼1.3 were observed during organic aerosol accumulation events (Sun et al., 2009). Sheesley et al. (2004) found that SOA, primarily biogenic-derived, contributed over 90 % of the total OC mass observed during the summer at the Seney National Wildlife Refuge in northern Michigan, located 120 km northwest of UMBS. Notably, ultrafine particle growth was observed at UMBS on 16 July during this high O∕C ratio spike (Gunsch et al., 2018a). The air arriving during this period was not under the influence of wildfires (Sect. 3.3) or urban areas (Sect. 3.4) and is therefore expected to be representative of remote background conditions.
3.3 Wildfire influence
From 13 to 15 July and 24 July midday through 25 July, the NOAA Hazard Mapping System (HMS) Smoke Product (Rolph et al., 2009) indicated that smoke plumes originating from wildfires within the Northwest Territories (Canada) directly influenced UMBS (Fig. 4). According to the Canadian Interagency Forest Fire Centre, over 5500 km2 of land burned within the Northwest Territories during July 2014 (CIFFC, 2014). Canadian wildfires are a major source of global PM2.5, with estimates of ∼1.6 Tg yr−1 emitted to the atmosphere (Wiedinmyer et al., 2006). Average PM2.5 number and mass concentrations during these two wildfire-influenced periods were statistically higher (t test, α=0.05) at 1400±800 particles cm−3 (range of 147–4832 particles cm−3, Fig. 1) and 5±3 µg m−3 (range of 1.3–10.5 µg m−3, Fig. 1), respectively, compared to the background period (Sect. 3.2). The particle number mode during wildfire influence was 80±46 nm, similar to the background air mass period (mode of 82±37 nm) (Fig. S3). Ozone was also elevated during 13–15 July reaching as high as 35 ppb, compared to an average of 10 ppb during the background period (Fig. 1). During these periods, the air masses did not pass over any major urban areas (Fig. S2), making ozone production within the smoke plume during transport the likely source (Jaffe and Wigder, 2012). Ozone did not increase during the 24 July smoke plume, staying near the average for the study (25±12 ppb) with a concentration of 26±3 ppb (Fig. 1). While an ozone increase is often observed for aged wildfire plumes, an increase does not always occur during wildfire influence, such as when low NOx levels within plumes, potentially due to smoldering combustion, limit the production of ozone (Jaffe and Wigder, 2012).
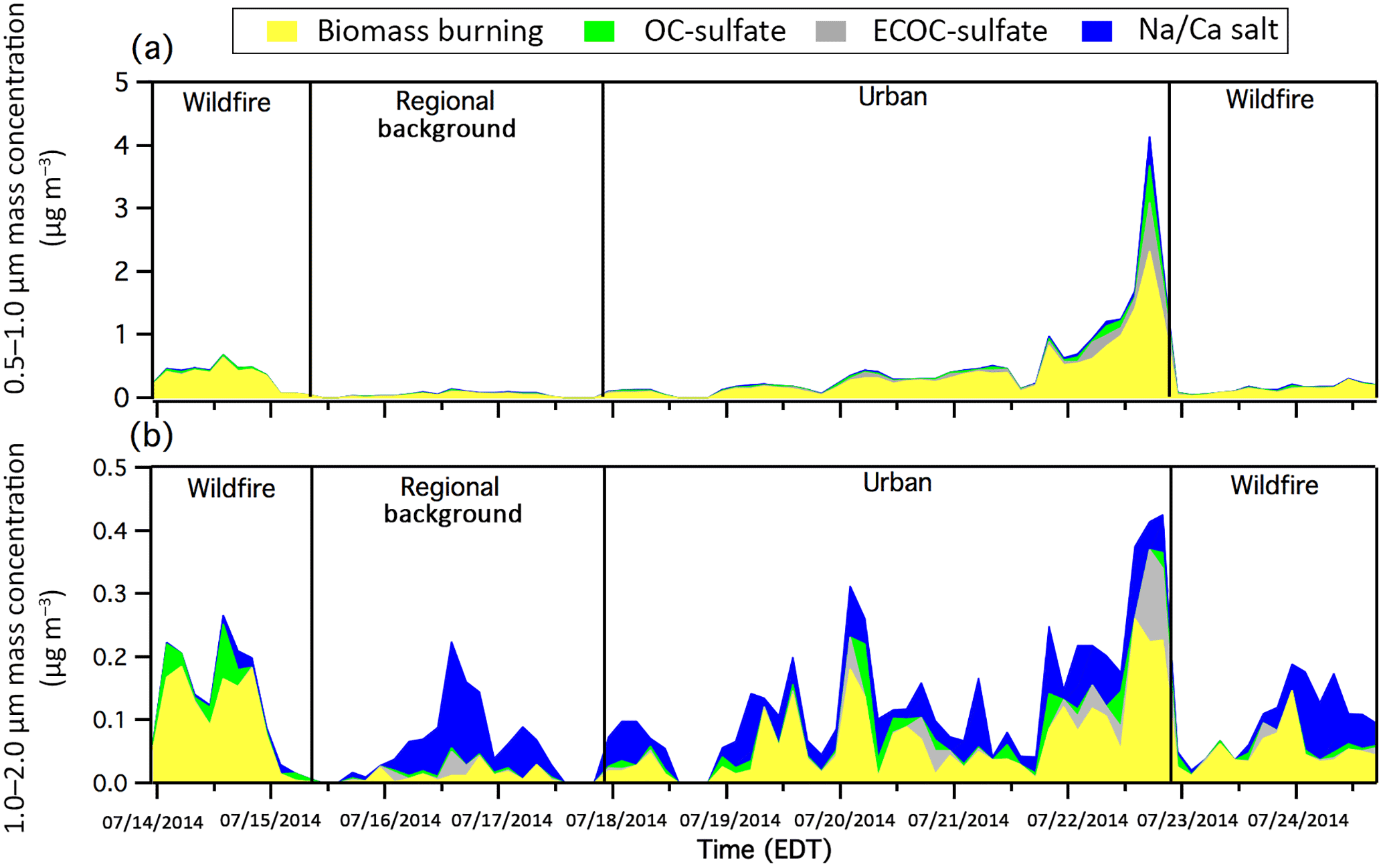
Figure 5The 3 h binned mass concentrations of (a) 0.5–1.0 µm and (b) 1.0–2.0 µm particle types, as measured with the ATOFMS. Gaps in the data correspond to periods when APS data were not available for scaling.
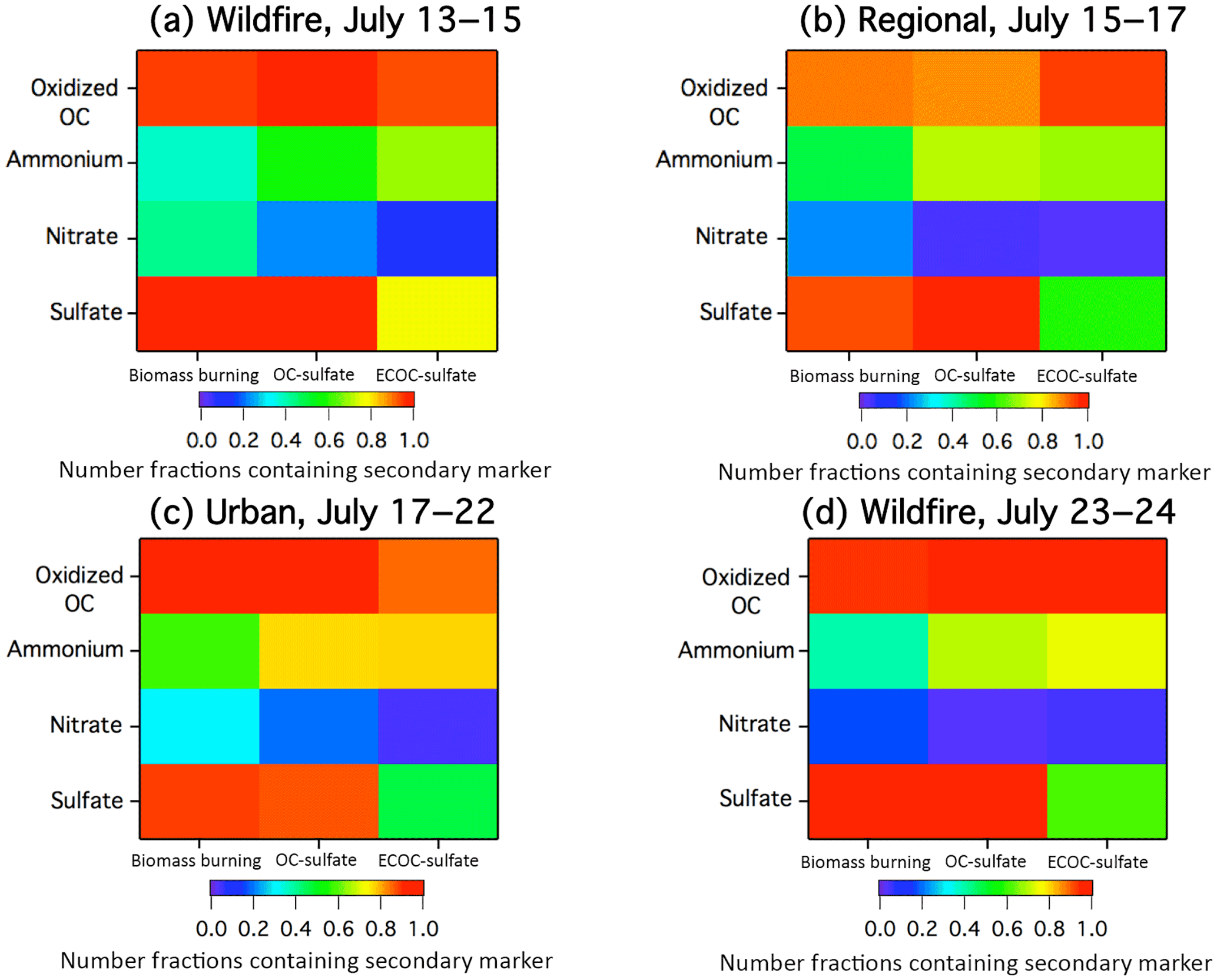
Figure 6Number fractions of individual biomass burning, OC-sulfate, and ECOC-sulfate particles that were internally mixed with secondary species, as determined by ATOFMS ion markers, including oxidized OC (C2H3O+, m∕z 43), ammonium (, m∕z 18), nitrate (, m∕z −46, and/or , m∕z −62), and sulfate (, m∕z −97). Since a given particle can contain more than one secondary species, the number fractions can add to greater than 1 for a given particle type. Chemical mixing states are provided here for the four air mass time periods: (a) wildfire influence from 13 to 15 July, (b) clean air from northern Canada from 15 to 17 July, (c) mix of wildfire and urban influences from 17 to 22 July, (d) wildfire influence from 23 to 24 July.
During the wildfire-influenced periods, 88±1 % of the measured 0.5–2.0 µm particles, by number, were biomass burning particles, with an average mass concentration of 0.42 µg m−3 (Fig. 5) and a maximum of 0.80 µg m−3 occurring during the early afternoon of 14 July when the heaviest wildfire smoke was reported by the NOAA smoke product (Fig. 4a). Minor contributions of OC-sulfate particles (8±1 % by number) were also measured. The OC-sulfate particle mass spectra (Fig. 2b) showed that 75±5 %, by number, contained potassium (K+, m∕z 39), suggesting that these were highly aged biomass burning particles coated by SOA such that the typical biomass burning mass spectral signature had been masked, as observed previously by Pratt and Prather (2009) using a thermodenuder. These OC-sulfate particles featured a dominant intense m∕z 43 (C2H3O+) ion peak, indicating that these particles were heavily coated with SOA. During the afternoon event on 24 July, PM1 organic mass concentrations measured with the HR-AMS nearly doubled from 2.5±0.1 µg m−3 before the event to 4.5±0.3 µg m−3 during the event (Fig. 3), accounting for ∼90 % of the total PM1 mass concentration. The HR-AMS O∕C ratio was 0.8 during wildfire periods, consistent with biomass burning particles heavily coated with SOA (Aiken et al., 2008), as also observed by 95±1 %, by number, of the biomass burning and OC-sulfate particles, measured with the ATOFMS during these periods, featured the oxidized OC ion marker (m∕z 43, C2H3O+) (Fig. 6). Freshly emitted biomass burning aerosol has an O∕C ratio of ∼0.2, which can increase to ∼0.6 in only a few hours as the emissions undergo photochemical aging and oxidized material condenses onto the particles (Grieshop et al., 2009; Pratt et al., 2011; Liu et al., 2016; Zhou et al., 2017). While AMS levoglucosan ion markers (m∕z 60 () and 73 () (Alfarra et al., 2007) were observed, consistent with the ATOFMS observation of biomass burning particles, the levels were low (Fig. S6), due to the expected degradation during atmospheric transport (Hennigan et al., 2010). The wildfire air masses measured during the present study were transported 48–72 h over Canadian forests, suggesting the accumulation of both biomass burning SOA and biogenic SOA. Previous studies conducted by Slowik et al. (2010) and Sheesley et al. (2004) observed monoterpene-derived SOA within summertime air masses passing over these forests. Therefore, it is likely that the oxidation (both gas and aqueous phase) of both biomass burning and biogenic VOCs (Lee et al., 2011), contributed to the observed elevated O∕C ratio of 0.8–1.1 at UMBS.
During transport of the biomass burning aerosols, accumulation of sulfate also occurred, with 97±1 %, by number, of biomass burning particles internally mixed with sulfate (m∕z −97, ) (Fig. 6). HR-AMS-measured PM1 sulfate also increased from less than 0.1 to 2 µg m−3 after midday 24 July (Fig. 3). SO2 has been previously shown to be emitted from wildfires (e.g., Burling et al., 2010; Stockwell et al., 2014), and increases in particulate sulfate mass have been observed during wildfire plume aging through cloud processing (DeBell et al., 2004; Pratt et al., 2010). In comparison, the HR-AMS measured limited amounts of PM1 ammonium (∼2 % of total mass, 0.2 µg m−3) during the wildfire event on 24 July (Fig. 3). However, ammonium was internally mixed in 38±2 %, by number, of biomass burning and 68±2 %, by number, of OC-sulfate particles (Fig. 6). This result indicates that while ammonium was present within many particles, it was a minor fraction of the particle mass. Nitrate was also internally mixed with 43±2 % of biomass burning particles, by number, and 17±2 %, by number, of OC-sulfate particles (Fig. 6), and the HR-AMS only measured ∼1 % of PM1 mass to be nitrate (0.06 µg m−3). Therefore, it is likely that the ammonium was present in the form of ammonium sulfate internally mixed with biomass burning and OC-sulfate particles.
3.4 Urban air mass influence
From 17 to 22 July, UMBS was influenced by air masses from the southwest, passing over the major metropolitan areas of Chicago and Milwaukee before arriving at the site (Fig. S2) after transport times of 24–36 h. Stagnant air (wind speeds of ∼2 m s−1) led to the buildup of the urban-influenced PM, which peaked on 22 July, as shown in Fig. 1. The passing of a cold front, along with precipitation and a change in wind direction, led to a sudden decrease in PM concentration late on 22 July (Fig. 1). The average ozone concentration was elevated at an average of 41±12 ppb similar to previous measurements by Cooper et al. (2001) at UMBS when under the direct influence of urban pollution (Fig. 1). The PM2.5 number and mass concentrations for this period were 2700±900 particles cm−3 (range of 414–6031 particles cm−3) and 14±8 µg m−3 (range of 2–43 µg m−3), respectively, the highest for the study (Fig. 1). The particle mode of 69±29 nm was also the smallest of the study (Fig. S3) due to contributions from combustion emissions, typically less than 50 nm (Seinfeld and Pandis, 2016), which likely grew to the observed sizes due to the condensation of secondary species during transport. A similar mode of 84±18 nm was observed by VanReken et al. (2015) at UMBS during summer 2009 urban air mass influence. VanReken et al. (2015) also previously observed the highest particle number concentrations (3000±1300 particles cm−3) at UMBS during the influence of southern air masses. Smoke influence was present during this period as shown by the NOAA smoke product (Fig. 4c). However, unlike during the previous periods, this smoke originated mainly from the southern United States and was from agricultural and forest fires (active agricultural fires were located in Missouri, with a forest fire in Arkansas). The HR-AMS showed levoglucosan ion markers (m∕z 60 () and 73 () (Alfarra et al., 2007) (Fig. S6), consistent with the satellite-observed smoke influence. Biomass burning particles measured with the ATOFMS steadily increased in mass concentration throughout this period (Fig. 5), with a notable spike in the mass concentration on 22 July observed in both the submicron (0.5–1.0 µm particles: 2.3 µg m−3) and supermicron (1.0–2.0 µm particles: 0.3 µg m−3) size ranges (Fig. 5). Overall, during urban influence, biomass burning particles accounted for 72±2 % of the particles, by number, and ∼30 % of the total mass concentration (Fig. 5). The biomass burning particles were aged, as shown by internal mixtures of sulfate (88±2 %, by number), oxidized OC (92±1 %, by number), ammonium (58±2 %, by number), and nitrate (30±2 %, by number) (Fig. 6). Notably, a higher number fraction of the biomass burning particles during this period were internally mixed with ammonium, compared to the biomass burning particles detected during the Canadian wildfire influence. The HR-AMS also measured the highest average ammonium mass concentration during this period of 1.6 µg m−3, accounting for 10 % of the total PM1 particle mass (Fig. 3). Agricultural activities, both crop and livestock, located to the south and southwest of the field site (Stephen and Aneja, 2008; Paulot et al., 2014) may be the source of the elevated ammonium levels.
ECOC-sulfate and OC-sulfate particles comprised the second most prominent particle types measured with the ATOFMS during this urban-influenced period at 12±1 % and 9±1 % of the submicron (0.5–1.0 µm) particles, by number, and an average of 0.08 and 0.03 µg m−3, respectively (Fig. 5). The influence of urban vehicular combustion resulted in the increased levels of measured ECOC-sulfate particles (Toner et al., 2006, 2008), compared to non-urban-influenced periods (2±1 % by number). HR-AMS PM1 mass concentrations (Fig. 3) showed increased organic mass during urban influence with an average mass concentration of 9.7 µg m−3 (Fig. 3), likely due to a mixture of biomass burning, anthropogenic, and biogenic organic aerosol. The average HR-AMS O∕C ratio during the urban period was the lowest of the study (0.78), likely due to increased contributions from hydrocarbon-like organic aerosol from urban vehicle combustion emissions (Aiken et al., 2008), in contrast to primarily oxidized organic aerosol during regional background periods (Jimenez et al., 2009). An increase in less-oxidized organic aerosol was similarly observed in rural Ontario when the site was influenced by urban air masses from Detroit, compared to remote air masses (Sjostedt et al., 2011). The ECOC-sulfate and OC-sulfate particles were highly aged, with ∼75 %, by number, of each particle type internally mixed with ammonium, consistent with particle aging during transport (Fig. 6c). Ammonium (1.6 µg m−3) and sulfate (4.9 µg m−3) comprised over 40 % of the total PM1 mass measured by the HR-AMS during this period, likely in the form of ammonium sulfate (Fig. 3). Urban-influenced air masses had the highest mass concentration of sulfate (up to 10 µg m−3) measured throughout the study. In contrast, there was little presence of nitrate internally mixed in the ECOC-sulfate (4±2 %, by number) and OC-sulfate (19±5 %, by number) particles (Fig. 6). Finally, while nitrate only comprised 1 % (0.2 µg m−3) of the total PM1 mass concentration during the urban air mass influence (Fig. 3), this was the highest nitrate contribution during the study, similar to previous rural eastern US studies (Jimenez et al., 2009).
Source apportionment of atmospheric particles in the summertime was conducted at the forested University of Michigan Biological Station, located in remote northern Michigan. The field site was impacted by air masses from three distinct areas: remote background, northwestern Canada, and southwestern urban areas. July 2014 was one of the most active burning seasons for the Northwest Territories in over 2 decades with a total of 10 643 km2 of land burned, significantly more than the 10-year (1944 km2) and 25-year (2423 km2) averages (CIFFC, 2014). The increased wildfire activity noticeably impacted northern Michigan, as the presence of biomass burning particles was ubiquitous throughout the study and made up the majority of measured particle number and mass concentrations. When air came from urban areas southwest of UMBS, aged biomass burning particles also dominated particle number concentrations due to wildfire influences from the southern United States. Due to the urban influence, these air masses had the highest mass contributions of sulfate (over 50 times the background) detected during the entire study. The accumulation of soluble secondary species, including sulfate and nitrate, increases the CCN ability of biomass burning particles (Furutani et al., 2008; Petters et al., 2009; J. Wang et al., 2010), illustrating the importance of transported wildfire emissions.
While biomass burning particles were the most dominant particle core detected, SOA was a major contributor to particle mass during the study. On average, the HR-AMS organic aerosol O∕C ratio was 0.84, indicative of highly oxidized OC (Aiken et al., 2008). During remote background periods, internal mixing of oxidized OC combined with the significant PM1 organic mass loading is indicative of the high mass loading of biogenic SOA in the forested region (Sheesley et al., 2004). During wildfire-influenced air masses, organics contributed ∼90 % to the PM1 mass, with SOA internally mixed with biomass burning and OC-sulfate particles, indicating that SOA from both biogenic VOC oxidation and wildfire combustion is a major source of OC in the region. The observed levels of biomass burning aerosol influence are attributed to the abnormally active Canadian wildfire season of 2014, compared to previous typical summers in northern Michigan with primarily biogenic SOA influence (Sheesley et al., 2004). Models underpredict OC in this region, and Jathar et al. (2014) indicates that on a national level, models predict biomass burning is the largest combustion contributor to SOA by mass, consistent with the significant influence of wildfires during this work.
Modeling studies have called for further investigations of wildfire emissions and areas they affect in order to reduce uncertainty within models due to limited data, particularly when modeling interactions between wildfire plumes and urban emissions. Wildfire plume ozone production can lead to areas far from the original source to be out of compliance with regulatory standards, demonstrating the importance to be able to accurately model ozone production (Hu et al., 2008; Jaffe and Wigder, 2012; Lu et al., 2016). Also, as described here, particles aged through transport show internal mixtures of nitrate, sulfate, and oxidized organics, which can lead to increased CCN activity (Furutani et al., 2008). With wildfires expected to increase in both intensity and frequency due to climate change (Gillett et al., 2004; Liu et al., 2010; Knorr et al., 2016; Veira et al., 2016), the contributions of long-range transported biomass burning emissions to the upper Midwest US atmosphere are expected to increase, such that air quality modeling efforts will need to supplement their existing emissions to account for the expected increase in wildfire emissions (Smith and Mueller, 2010).
Data are available at the University of Michigan Biological Station data archive (Gunsch et al., 2018b) and by contacting the corresponding authors.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-3701-2018-supplement.
The authors declare that they have no conflict of interest.
Funding for the UMBS study was provided by the University of Michigan MCubed
Program and UMBS graduate fellowships for Matthew J. Gunsch, Nathaniel W. May, and Daniel J. Gardner.
We also thank the Pratt and Ault groups for assistance on the field study.
Support for Timothy M. VanReken and Miao Wen was provided by the US Department of
Energy Early Career Research Program (award no. SC0003899). Jennifer Dean
(Washington State University) is thanked for assistance during the field
study. Donna Sueper (Aerodyne) is thanked for assistance with HR-AMS data
processing. The authors gratefully acknowledge the NOAA Air Resources
Laboratory for the provision of the HYSPLIT transport and dispersion
model and READY website (http://www.ready.noaa.gov) used in this
publication. The authors also gratefully acknowledge the NOAA Office of
Satellite and Product Operations for the use of the Hazard Mapping System
Smoke Product
(http://www.ospo.noaa.gov/Products/land/hms.html).
Edited by: Sally E. Pusede
Reviewed by: two anonymous referees
Aiken, A. C., Decarlo, P. F., Kroll, J. H., Worsnop, D. R., Huffman, J. A., Docherty, K. S., Ulbrich, I. M., Mohr, C., Kimmel, J. R., and Sueper, D.: O∕C and OM∕OC ratios of primary, secondary, and ambient organic aerosols with high-resolution time-of-flight aerosol mass spectrometry, Environ. Sci. Technol., 42, 4478–4485, 2008.
Alfarra, M. R., Prevot, A. S., Szidat, S., Sandradewi, J., Weimer, S., Lanz, V. A., Schreiber, D., Mohr, M., and Baltensperger, U.: Identification of the mass spectral signature of organic aerosols from wood burning emissions, Environ. Sci. Technol., 41, 5770–5777, 2007.
Allan, J. D., Jimenez, J. L., Williams, P. I., Alfarra, M. R., Bower, K. N., Jayne, J. T., Coe, H., and Worsnop, D. R.: Quantitative sampling using an Aerodyne aerosol mass spectrometer 1. Techniques of data interpretation and error analysis, J. Geophys. Res.-Atmos., 108, 4090, https://doi.org/10.1029/2002JD002359, 2003.
Allan, J. D., Delia, A. E., Coe, H., Bower, K. N., Alfarra, M. R., Jimenez, J. L., Middlebrook, A. M., Drewnick, F., Onasch, T. B., and Canagaratna, M. R.: A generalised method for the extraction of chemically resolved mass spectra from Aerodyne aerosol mass spectrometer data, J. Aerosol Sci., 35, 909–922, 2004.
Ault, A. P., Williams, C. R., White, A. B., Neiman, P. J., Creamean, J. M., Gaston, C. J., Ralph, F. M., and Prather, K. A.: Detection of Asian dust in California orographic precipitation, J. Geophys. Res.-Atmos., 116, D16205, https://doi.org/10.1029/2010JD015351, 2011.
Axson, J. L., May, N. W., Colón-Bernal, I. D., Pratt, K. A., and Ault, A. P.: Lake spray aerosol: a chemical signature from individual ambient particles, Environ. Sci. Technol., 50, 9835–9845, 2016.
Bauer, S. E., Ault, A., and Prather, K. A.: Evaluation of aerosol mixing state classes in the GISS modelE-MATRIX climate model using single-particle mass spectrometry measurements, J. Geophys. Res.-Atmos., 118, 9834–9844, 2013.
Brook, R. D., Franklin, B., Cascio, W., Hong, Y., Howard, G., Lipsett, M., Luepker, R., Mittleman, M., Samet, J., and Smith, S. C.: Air pollution and cardiovascular disease, Circulation, 109, 2655–2671, 2004.
Bullard, R. L., Singh, A., Anderson, S. M., Lehmann, C. M., and Stanier, C. O.: 10-Month characterization of the aerosol number size distribution and related air quality and meteorology at the Bondville, IL Midwestern background site, Atmos. Environ., 174, 348–361, 2017.
Burling, I. R., Yokelson, R. J., Griffith, D. W. T., Johnson, T. J., Veres, P., Roberts, J. M., Warneke, C., Urbanski, S. P., Reardon, J., Weise, D. R., Hao, W. M., and de Gouw, J.: Laboratory measurements of trace gas emissions from biomass burning of fuel types from the southeastern and southwestern United States, Atmos. Chem. Phys., 10, 11115–11130, https://doi.org/10.5194/acp-10-11115-2010, 2010.
Calvo, A., Alves, C., Castro, A., Pont, V., Vicente, A., and Fraile, R.: Research on aerosol sources and chemical composition: past, current and emerging issues, Atmos. Res., 120, 1–28, 2013.
Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R.: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 15, 253–272, https://doi.org/10.5194/acp-15-253-2015, 2015.
Carlton, A. G., Pinder, R. W., Bhave, P. V., and Pouliot, G. A.: To what extent can biogenic SOA be controlled?, Environ. Sci. Technol., 44, 3376–3380, 2010.
Carroll, M. A., Bertman, S. B., and Shepson, P. B.: Overview of the Program for Research on Oxidants: Photochemistry, Emissions, and Transport (PROPHET) summer 1998 measurements intensive, J. Geophys. Res.-Atmos., 106, 24275–24288, 2001.
CIFFC: Canadian Interagency Forest Fire Centre, National Wildland Fire Situation Report: http://www.ciffc.ca, last access: July 2016.
Colarco, P., Schoeberl, M., Doddridge, B., Marufu, L., Torres, O., and Welton, E.: Transport of smoke from Canadian forest fires to the surface near Washington, DC: injection height, entrainment, and optical properties, J. Geophys. Res.-Atmos., 109, D06203, https://doi.org/10.1029/2003JD004248, 2004.
Cooper, O., Moody, J., Thornberry, T., Town, M., and Carroll, M.: PROPHET 1998 meteorological overview and air-mass classification, J. Geophys. Res.-Atmos., 106, 24289–24299, 2001.
Creamean, J. M., Suski, K. J., Rosenfeld, D., Cazorla, A., DeMott, P. J., Sullivan, R. C., White, A. B., Ralph, F. M., Minnis, P., and Comstock, J. M.: Dust and biological aerosols from the Sahara and Asia influence precipitation in the western US, Science, 339, 1572–1578, 2013.
Dall'Osto, M., Beddows, D., Kinnersley, R. P., Harrison, R. M., Donovan, R. J., and Heal, M. R.: Characterization of individual airborne particles by using aerosol time-of-flight mass spectrometry at Mace Head, Ireland, J. Geophys. Res.-Atmos., 109, D21302, https://doi.org/10.1029/2004JD004747, 2004.
DeBell, L. J., Talbot, R. W., Dibb, J. E., Munger, J. W., Fischer, E. V., and Frolking, S. E.: A major regional air pollution event in the northeastern United States caused by extensive forest fires in Quebec, Canada, J. Geophys. Res.-Atmos., 109, D19305, https://doi.org/10.1029/2004JD004840, 2004.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., and Docherty, K. S.: Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer, Anal. Chem., 78, 8281–8289, 2006.
Dempsey, F.: Forest fire effects on air quality in Ontario: evaluation of several recent examples, B. Am. Meteorol. Soc., 94, 1059–1064, 2013.
Dreessen, J., Sullivan, J., and Delgado, R.: Observations and impacts of transported canadian wildfire smoke on ozone and aerosol air quality in the Maryland region on 9–12 June, 2015, J. J Air Waste Manage., 66, 842–862, 2016.
Dutkiewicz, V. A., Husain, L., Roychowdhury, U. K., and Demerjian, K. L.: Impact of Canadian wildfire smoke on air quality at two rural sites in NY State, Atmos. Environ., 45, 2028–2033, 2011.
Emanuelsson, E. U., Hallquist, M., Kristensen, K., Glasius, M., Bohn, B., Fuchs, H., Kammer, B., Kiendler-Scharr, A., Nehr, S., Rubach, F., Tillmann, R., Wahner, A., Wu, H.-C., and Mentel, Th. F.: Formation of anthropogenic secondary organic aerosol (SOA) and its influence on biogenic SOA properties, Atmos. Chem. Phys., 13, 2837–2855, https://doi.org/10.5194/acp-13-2837-2013, 2013.
Farmer, D., Matsunaga, A., Docherty, K., Surratt, J., Seinfeld, J., Ziemann, P., and Jimenez, J.: Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry, P. Natl. Acad. Sci. USA, 107, 6670–6675, 2010.
Fierce, L., Bond, T. C., Bauer, S. E., Mena, F., and Riemer, N.: Black carbon absorption at the global scale is affected by particle-scale diversity in composition, Nat. Commun., 7, 12361, https://doi.org/10.1038/ncomms12361, 2016.
Fitzgerald, E., Ault, A. P., Zauscher, M. D., Mayol-Bracero, O. L., and Prather, K. A.: Comparison of the mixing state of long-range transported Asian and African mineral dust, Atmos. Environ., 115, 19–25, 2015.
Forster, C., Wandinger, U., Wotawa, G., James, P., Mattis, I., Althausen, D., Simmonds, P., O'Doherty, S., Jennings, S. G., and Kleefeld, C.: Transport of boreal forest fire emissions from Canada to Europe, J. Geophys. Res.-Atmos., 106, 22887–22906, 2001.
Furutani, H., Dall'osto, M., Roberts, G. C., and Prather, K. A.: Assessment of the relative importance of atmospheric aging on CCN activity derived from field observations, Atmos. Environ., 42, 3130–3142, 2008.
Gard, E., Mayer, J. E., Morrical, B. D., Dienes, T., Fergenson, D. P., and Prather, K. A.: Real-time analysis of individual atmospheric aerosol particles: design and performance of a portable ATOFMS, Anal. Chem., 69, 4083–4091, 1997.
Gillett, N., Weaver, A., Zwiers, F., and Flannigan, M.: Detecting the effect of climate change on Canadian forest fires, Geophys. Res. Lett., 31, L18211, https://doi.org/10.1029/2004GL020876, 2004.
Grieshop, A. P., Donahue, N. M., and Robinson, A. L.: Laboratory investigation of photochemical oxidation of organic aerosol from wood fires 2: analysis of aerosol mass spectrometer data, Atmos. Chem. Phys., 9, 2227–2240, https://doi.org/10.5194/acp-9-2227-2009, 2009.
Gunsch, M. J., Schmidt, S., Gardner, D. J., Bondy, A. L., May, N., Bertman, S. B., Pratt, K. A., and Ault, A. P.: Particle growth in an isoprene-rich forest: influences of urban, wildfire, and biogenic precursors, Atmos. Environ., 178, 255–264, https://doi.org/10.1016/j.atmosenv.2018.01.058, 2018a.
Gunsch, M. J., Pratt, K. A., Ault, A. P., and VanReken, T. M.: UMBS: PROPHET Aerosol and Ozone Data, July 2014, Environmental Data Initiative, https://doi.org/10.6073/pasta/b0943aacce8ad7dea732e2442d22ac53, 2018b.
Hand, J. L., Copeland, S. A., Day, D. E., Dillner, A. M., Indresand, H., Malm, W. C., McDade, C. E., Moore, C. T., Pitchford, M. L., Schichtel, B. A., and Watson, J. G.: Spatial and Seasonal Patterns and Temporal Variability of Haze and its Constituents in the United States: Report V, available at: http://vista.cira.colostate.edu/improve/wp-content/uploads/2016/08/IMPROVE_V_FullReport.pdf (last access: August 2017), 2011.
Hennigan, C. J., Sullivan, A. P., Collett, J. L., and Robinson, A. L.: Levoglucosan stability in biomass burning particles exposed to hydroxyl radicals, Geophys. Res. Lett., 37, L09806, https://doi.org/10.1029/2010GL043088, 2010.
Hu, Y., Odman, M. T., Chang, M. E., Jackson, W., Lee, S., Edgerton, E. S., Baumann, K., and Russell, A. G.: Simulation of air quality impacts from prescribed fires on an urban area, Environ. Sci. Technol., 42, 3676–3682, 2008.
Hudson, P. K., Murphy, D. M., Cziczo, D. J., Thomson, D. S., De Gouw, J. A., Warneke, C., Holloway, J., Jost, H. J., and Hübler, G.: Biomass-burning particle measurements: characteristic composition and chemical processing, J. Geophys. Res.-Atmos., 109, D23S27, https://doi.org/10.1029/2003JD004398, 2004.
IPCC: IPCC, 2013: climate change 2013: the physical science basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Stocker, T., Qin, D., Plattner, G., Tignor, M., Allen, S., Boschung, J., Nauels, A., Xia, Y., Bex, B., and Midgley, B., Cambridge University Press, Cambridge, UK and New York, NY, USA, 2013.
Jaffe, D. A. and Wigder, N. L.: Ozone production from wildfires: a critical review, Atmos. Environ., 51, 1–10, 2012.
Jathar, S. H., Gordon, T. D., Hennigan, C. J., Pye, H. O., Pouliot, G., Adams, P. J., Donahue, N. M., and Robinson, A. L.: Unspeciated organic emissions from combustion sources and their influence on the secondary organic aerosol budget in the United States, P. Natl. Acad. Sci. USA, 111, 10473–10478, 2014.
Jayne, J. T., Leard, D. C., Zhang, X., Davidovits, P., Smith, K. A., Kolb, C. E., and Worsnop, D. R.: Development of an aerosol mass spectrometer for size and composition analysis of submicron particles, Aerosol Sci. Tech., 33, 49–70, 2000.
Jeong, C.-H., McGuire, M. L., Godri, K. J., Slowik, J. G., Rehbein, P. J. G., and Evans, G. J.: Quantification of aerosol chemical composition using continuous single particle measurements, Atmos. Chem. Phys., 11, 7027–7044, https://doi.org/10.5194/acp-11-7027-2011, 2011.
Jimenez, J. and DeCarlo, P.: Field ToF-AMS Operation: http://cires1.colorado.edu/jimenez-group/wiki/index.php/Field_ToF-AMS_Operation, last access: September 2017.
Jimenez, J., Canagaratna, M., Donahue, N., Prevot, A., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., and Ng, N.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525–1529, 2009.
Jimenez, J. L., Jayne, J. T., Shi, Q., Kolb, C. E., Worsnop, D. R., Yourshaw, I., Seinfeld, J. H., Flagan, R. C., Zhang, X., and Smith, K. A.: Ambient aerosol sampling using the aerodyne aerosol mass spectrometer, J. Geophys. Res.-Atmos., 108, 8425, 2003.
Kang, C.-M., Gold, D., and Koutrakis, P.: Downwind O3 and PM2.5 speciation during the wildfires in 2002 and 2010, Atmos. Environ., 95, 511–519, 2014.
Khlystov, A., Stanier, C., and Pandis, S.: An algorithm for combining electrical mobility and aerodynamic size distributions data when measuring ambient aerosol, Aerosol Sci. Tech., 38, 229–238, 2004.
Kim, E., Hopke, P. K., Kenski, D. M., and Koerber, M.: Sources of fine particles in a rural midwestern US area, Environ. Sci. Technol., 39, 4953–4960, 2005.
Kim, M., Deshpande, S. R., and Crist, K. C.: Source apportionment of fine particulate matter (PM2.5) at a rural Ohio River Valley site, Atmos. Environ., 41, 9231–9243, 2007.
Knorr, W., Jiang, L., and Arneth, A.: Climate, CO2 and human population impacts on global wildfire emissions, Biogeosciences, 13, 267–282, https://doi.org/10.5194/bg-13-267-2016, 2016.
Kundu, S. and Stone, E. A.: Composition and sources of fine particulate matter across urban and rural sites in the Midwestern United States, Env. Sci. Process. Impact, 16, 1360–1370, 2014.
Lee, A. K., Herckes, P., Leaitch, W., Macdonald, A., and Abbatt, J.: Aqueous OH oxidation of ambient organic aerosol and cloud water organics: formation of highly oxidized products, Geophys. Res. Lett., 38, L11805, https://doi.org/10.1029/2011GL047439, 2011.
Liu, X., Zhang, Y., Huey, L., Yokelson, R., Wang, Y., Jimenez, J., Campuzano-Jost, P., Beyersdorf, A., Blake, D., and Choi, Y.: Agricultural fires in the southeastern US during SEAC4RS: emissions of trace gases and particles and evolution of ozone, reactive nitrogen, and organic aerosol, J. Geophys. Res.-Atmos., 121, 7383–7414, 2016.
Liu, Y., Stanturf, J., and Goodrick, S.: Trends in global wildfire potential in a changing climate, Forest. Ecol. Manag., 259, 685–697, 2010.
Lu, X., Zhang, L., Yue, X., Zhang, J., Jaffe, D. A., Stohl, A., Zhao, Y., and Shao, J.: Wildfire influences on the variability and trend of summer surface ozone in the mountainous western United States, Atmos. Chem. Phys., 16, 14687–14702, https://doi.org/10.5194/acp-16-14687-2016, 2016.
Matsui, H., Koike, M., Kondo, Y., Moteki, N., Fast, J. D., and Zaveri, R. A.: Development and validation of a black carbon mixing state resolved three-dimensional model: aging processes and radiative impact, J. Geophys. Res.-Atmos., 118, 2304–2326, 2013.
May, N. W., Olson, N. E., Panas, M., Axson, J. L., Tirella, P. S., Kirpes, R. M., Craig, R. L., Gunsch, M. J., China, S., Laskin, A., Ault, A. P., and Pratt, K. A.: Aerosol emissions from Great Lakes harmful algal blooms, Environ. Sci. Technol., 52, 397–405, https://doi.org/10.1021/acs.est.7b03609, 2018.
Middlebrook, A. M., Bahreini, R., Jimenez, J. L., and Canagaratna, M. R.: Evaluation of composition-dependent collection efficiencies for the Aerodyne aerosol mass spectrometer using field data, Aerosol Sci. Tech., 46, 258–271, 2012.
Miller, D. J., Sun, K., Zondlo, M. A., Kanter, D., Dubovik, O., Welton, E. J., Winker, D. M., and Ginoux, P.: Assessing boreal forest fire smoke aerosol impacts on US air quality: a case study using multiple data sets, J. Geophys. Res.-Atmos., 116, D22209, https://doi.org/10.1029/2011JD016170, 2011.
Moffet, R. C. and Prather, K. A.: In-situ measurements of the mixing state and optical properties of soot with implications for radiative forcing estimates, P. Natl. Acad. Sci. USA, 106, 11872–11877, 2009.
Moffet, R. C., Qin, X., Rebotier, T., Furutani, H., and Prather, K. A.: Chemically segregated optical and microphysical properties of ambient aerosols measured in a single-particle mass spectrometer, J. Geophys. Res.-Atmos., 113, D12213, https://doi.org/10.1029/2007JD009393, 2008.
Moffet, R. C., Henn, T. R., Tivanski, A. V., Hopkins, R. J., Desyaterik, Y., Kilcoyne, A. L. D., Tyliszczak, T., Fast, J., Barnard, J., Shutthanandan, V., Cliff, S. S., Perry, K. D., Laskin, A., and Gilles, M. K.: Microscopic characterization of carbonaceous aerosol particle aging in the outflow from Mexico City, Atmos. Chem. Phys., 10, 961–976, https://doi.org/10.5194/acp-10-961-2010, 2010.
Müller, D., Mattis, I., Wandinger, U., Ansmann, A., Althausen, D., and Stohl, A.: Raman lidar observations of aged Siberian and Canadian forest fire smoke in the free troposphere over Germany in 2003: microphysical particle characterization, J. Geophys. Res.-Atmos., 110, D17201, https://doi.org/10.1029/2004JD005756, 2005.
National Research Council, and National Academies: Global Sources of Local Pollution: an Assessment of Long-Range Transport of Key Air Pollutants to and from the United States, National Academies Press, Washington, D.C., XIII, 234 p., 2010.
Pastor, S. H., Allen, J. O., Hughes, L. S., Bhave, P., Cass, G. R., and Prather, K. A.: Ambient single particle analysis in Riverside, California by aerosol time-of-flight mass spectrometry during the SCOS97-NARSTO, Atmos. Environ., 37, 239–258, 2003.
Paulot, F., Jacob, D. J., Pinder, R., Bash, J., Travis, K., and Henze, D.: Ammonia emissions in the United States, European Union, and China derived by high-resolution inversion of ammonium wet deposition data: interpretation with a new agricultural emissions inventory (MASAGE_NH3), J. Geophys. Res.-Atmos., 119, 4343–4364, 2014.
Petters, M. D., Carrico, C. M., Kreidenweis, S. M., Prenni, A. J., DeMott, P. J., Collett, J. L., and Moosmueller, H.: Cloud condensation nucleation activity of biomass burning aerosol, J. Geophys. Res.-Atmos., 114, D22205, https://doi.org/10.1029/2009JD012353, 2009.
Pope, C. A. and Dockery, D. W.: Health effects of fine particulate air pollution: lines that connect, J. Air Waste Manage., 56, 709–742, 2006.
Pöschl, U.: Atmospheric aerosols: composition, transformation, climate and health effects, Angew. Chem. Int. Edit., 44, 7520–7540, 2005.
Pöschl, U. and Shiraiwa, M.: Multiphase chemistry at the atmosphere–biosphere interface influencing climate and public health in the anthropocene, Chem. Rev., 115, 4440–4475, 2015.
Pratt, K. A. and Prather, K. A.: Real-time, single-particle volatility, size, and chemical composition measurements of aged urban aerosols, Environ. Sci. Technol., 43, 8276–8282, 2009.
Pratt, K. A. and Prather, K. A.: Mass spectrometry of atmospheric aerosols – Recent developments and applications. Part II: On-line mass spectrometry techniques, Mass Spectrom. Rev., 31, 17–48, 2012.
Pratt, K. A., Heymsfield, A. J., Twohy, C. H., Murphy, S. M., DeMott, P. J., Hudson, J. G., Subramanian, R., Wang, Z., Seinfeld, J. H., and Prather, K. A.: In situ chemical characterization of aged biomass-burning aerosols impacting cold wave clouds, J. Atmos. Sci., 67, 2451–2468, 2010.
Pratt, K. A., Murphy, S. M., Subramanian, R., DeMott, P. J., Kok, G. L., Campos, T., Rogers, D. C., Prenni, A. J., Heymsfield, A. J., Seinfeld, J. H., and Prather, K. A.: Flight-based chemical characterization of biomass burning aerosols within two prescribed burn smoke plumes, Atmos. Chem. Phys., 11, 12549–12565, https://doi.org/10.5194/acp-11-12549-2011, 2011.
Qin, X., Bhave, P. V., and Prather, K. A.: Comparison of two methods for obtaining quantitative mass concentrations from aerosol time-of-flight mass spectrometry measurements, Anal. Chem., 78, 6169–6178, 2006.
Qin, X., Pratt, K. A., Shields, L. G., Toner, S. M., and Prather, K. A.: Seasonal comparisons of single-particle chemical mixing state in Riverside, CA, Atmos. Environ., 59, 587–596, 2012.
Raatikainen, T., Vaattovaara, P., Tiitta, P., Miettinen, P., Rautiainen, J., Ehn, M., Kulmala, M., Laaksonen, A., and Worsnop, D. R.: Physicochemical properties and origin of organic groups detected in boreal forest using an aerosol mass spectrometer, Atmos. Chem. Phys., 10, 2063–2077, https://doi.org/10.5194/acp-10-2063-2010, 2010.
Rattanavaraha, W., Chu, K., Budisulistiorini, S. H., Riva, M., Lin, Y.-H., Edgerton, E. S., Baumann, K., Shaw, S. L., Guo, H., King, L., Weber, R. J., Neff, M. E., Stone, E. A., Offenberg, J. H., Zhang, Z., Gold, A., and Surratt, J. D.: Assessing the impact of anthropogenic pollution on isoprene-derived secondary organic aerosol formation in PM2.5 collected from the Birmingham, Alabama, ground site during the 2013 Southern Oxidant and Aerosol Study, Atmos. Chem. Phys., 16, 4897–4914, https://doi.org/10.5194/acp-16-4897-2016, 2016.
Riemer, N. and West, M.: Quantifying aerosol mixing state with entropy and diversity measures, Atmos. Chem. Phys., 13, 11423–11439, https://doi.org/10.5194/acp-13-11423-2013, 2013.
Rolph, G. D., Draxler, R. R., Stein, A. F., Taylor, A., Ruminski, M. G., Kondragunta, S., Zeng, J., Huang, H.-C., Manikin, G., and McQueen, J. T.: Description and verification of the NOAA smoke forecasting system: the 2007 fire season, Weather Forecast., 24, 361–378, 2009.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric Chemistry Physics: From Air Pollution to Climate Change, John Wiley & Sons, Hoboken, New Jersey, 2016.
Sheesley, R. J., Schauer, J. J., Bean, E., and Kenski, D.: Trends in secondary organic aerosol at a remote site in Michigan's upper peninsula, Environ. Sci. Technol., 38, 6491–6500, 2004.
Silva, P. J., Liu, D.-Y., Noble, C. A., and Prather, K. A.: Size and chemical characterization of individual particles resulting from biomass burning of local Southern California species, Environ. Sci. Technol., 33, 3068–3076, 1999.
Sjostedt, S. J., Slowik, J. G., Brook, J. R., Chang, R. Y.-W., Mihele, C., Stroud, C. A., Vlasenko, A., and Abbatt, J. P. D.: Diurnally resolved particulate and VOC measurements at a rural site: indication of significant biogenic secondary organic aerosol formation, Atmos. Chem. Phys., 11, 5745–5760, https://doi.org/10.5194/acp-11-5745-2011, 2011.
Slowik, J. G., Stroud, C., Bottenheim, J. W., Brickell, P. C., Chang, R. Y.-W., Liggio, J., Makar, P. A., Martin, R. V., Moran, M. D., Shantz, N. C., Sjostedt, S. J., van Donkelaar, A., Vlasenko, A., Wiebe, H. A., Xia, A. G., Zhang, J., Leaitch, W. R., and Abbatt, J. P. D.: Characterization of a large biogenic secondary organic aerosol event from eastern Canadian forests, Atmos. Chem. Phys., 10, 2825–2845, https://doi.org/10.5194/acp-10-2825-2010, 2010.
Slowik, J. G., Brook, J., Chang, R. Y.-W., Evans, G. J., Hayden, K., Jeong, C.-H., Li, S.-M., Liggio, J., Liu, P. S. K., McGuire, M., Mihele, C., Sjostedt, S., Vlasenko, A., and Abbatt, J. P. D.: Photochemical processing of organic aerosol at nearby continental sites: contrast between urban plumes and regional aerosol, Atmos. Chem. Phys., 11, 2991–3006, https://doi.org/10.5194/acp-11-2991-2011, 2011.
Smith, S. N. and Mueller, S. F.: Modeling natural emissions in the Community Multiscale Air Quality (CMAQ) Model–I: building an emissions data base, Atmos. Chem. Phys., 10, 4931–4952, https://doi.org/10.5194/acp-10-4931-2010, 2010.
Song, X.-H., Hopke, P. K., Fergenson, D. P., and Prather, K. A.: Classification of single particles analyzed by ATOFMS using an artificial neural network, ART-2A, Anal. Chem., 71, 860–865, 1999.
Spencer, M. T., Shields, L. G., and Prather, K. A.: Simultaneous measurement of the effective density and chemical composition of ambient aerosol particles, Environ. Sci. Technol., 41, 1303–1309, 2007.
Stephen, K. and Aneja, V. P.: Trends in agricultural ammonia emissions and ammonium concentrations in precipitation over the Southeast and Midwest United States, Atmos. Environ., 42, 3238–3252, 2008.
Stockwell, C. E., Yokelson, R. J., Kreidenweis, S. M., Robinson, A. L., DeMott, P. J., Sullivan, R. C., Reardon, J., Ryan, K. C., Griffith, D. W. T., and Stevens, L.: Trace gas emissions from combustion of peat, crop residue, domestic biofuels, grasses, and other fuels: configuration and Fourier transform infrared (FTIR) component of the fourth Fire Lab at Missoula Experiment (FLAME-4), Atmos. Chem. Phys., 14, 9727–9754, https://doi.org/10.5194/acp-14-9727-2014, 2014.
Su, Y., Sipin, M. F., Furutani, H., and Prather, K. A.: Development and characterization of an aerosol time-of-flight mass spectrometer with increased detection efficiency, Anal. Chem., 76, 712–719, 2004.
Sueper, D.: ToF-AMS analysis software, available at: http://cires1.colorado.edu/jimenez-group/ToFAMSResources/ToFSoftware/index.html (last access: August 2017), 2010.
Sullivan, R. C., Guazzotti, S. A., Sodeman, D. A., and Prather, K. A.: Direct observations of the atmospheric processing of Asian mineral dust, Atmos. Chem. Phys., 7, 1213–1236, https://doi.org/10.5194/acp-7-1213-2007, 2007.
Sun, Y., Zhang, Q., Macdonald, A. M., Hayden, K., Li, S. M., Liggio, J., Liu, P. S. K., Anlauf, K. G., Leaitch, W. R., Steffen, A., Cubison, M., Worsnop, D. R., van Donkelaar, A., and Martin, R. V.: Size-resolved aerosol chemistry on Whistler Mountain, Canada with a high-resolution aerosol mass spectrometer during INTEX-B, Atmos. Chem. Phys., 9, 3095–3111, https://doi.org/10.5194/acp-9-3095-2009, 2009.
Toner, S. M., Sodeman, D. A., and Prather, K. A.: Single particle characterization of ultrafine and accumulation mode particles from heavy duty diesel vehicles using aerosol time-of-flight mass spectrometry, Environ. Sci. Technol., 40, 3912–3921, 2006.
Toner, S. M., Shields, L. G., Sodeman, D. A., and Prather, K. A.: Using mass spectral source signatures to apportion exhaust particles from gasoline and diesel powered vehicles in a freeway study using UF-ATOFMS, Atmos. Environ., 42, 568–581, 2008.
Uno, I., Eguchi, K., Yumimoto, K., Takemura, T., Shimizu, A., Uematsu, M., Liu, Z., Wang, Z., Hara, Y., and Sugimoto, N.: Asian dust transported one full circuit around the globe, Nat. Geosci., 2, 557–560, 2009.
VanReken, T., Mwaniki, G., Wallace, H., Pressley, S., Erickson, M., Jobson, B., and Lamb, B.: Influence of air mass origin on aerosol properties at a remote Michigan forest site, Atmos. Environ., 107, 35–43, 2015.
Veira, A., Lasslop, G., and Kloster, S.: Wildfires in a warmer climate: emission fluxes, emission heights, and black carbon concentrations in 2090–2099, J. Geophys. Res.-Atmos., 121, 3195–3223, 2016.
Wang, J., Cubison, M. J., Aiken, A. C., Jimenez, J. L., and Collins, D. R.: The importance of aerosol mixing state and size-resolved composition on CCN concentration and the variation of the importance with atmospheric aging of aerosols, Atmos. Chem. Phys., 10, 7267–7283, https://doi.org/10.5194/acp-10-7267-2010, 2010.
Wang, Y., Huang, J., Zananski, T. J., Hopke, P. K., and Holsen, T. M.: Impacts of the Canadian forest fires on atmospheric mercury and carbonaceous particles in northern New York, Environ. Sci. Technol., 44, 8435–8440, 2010.
Wiedinmyer, C., Quayle, B., Geron, C., Belote, A., McKenzie, D., Zhang, X., O'Neill, S., and Wynne, K. K.: Estimating emissions from fires in North America for air quality modeling, Atmos. Environ., 40, 3419–3432, 2006.
Xu, L., Guo, H., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., and Knote, C.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, 37–42, 2015.
Zhang, Y., Sheesley, R. J., Schauer, J. J., Lewandowski, M., Jaoui, M., Offenberg, J. H., Kleindienst, T. E., and Edney, E. O.: Source apportionment of primary and secondary organic aerosols using positive matrix factorization (PMF) of molecular markers, Atmos. Environ., 43, 5567–5574, 2009.
Zhou, S., Collier, S., Jaffe, D. A., Briggs, N. L., Hee, J., Sedlacek III, A. J., Kleinman, L., Onasch, T. B., and Zhang, Q.: Regional influence of wildfires on aerosol chemistry in the western US and insights into atmospheric aging of biomass burning organic aerosol, Atmos. Chem. Phys., 17, 2477–2493, https://doi.org/10.5194/acp-17-2477-2017, 2017.