the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 04 Sep 2018
| 04 Sep 2018
Biomass burning emission disturbances of isoprene oxidation in a tropical forest
Fernando Santos
Karla Longo
Alex Guenther
Saewung Kim
Dave Oram
Grant Forster
James Lee
James Hopkins
Joel Brito
Saulo Freitas
We present a characterization of the chemical composition of the atmosphere of the Brazilian Amazon rainforest based on trace gas measurements carried out during the South AMerican Biomass Burning Analysis (SAMBBA) airborne experiment in September 2012. We analyzed the observations of primary biomass burning emission tracers, i.e., carbon monoxide (CO), nitrogen oxides (NOx), ozone (O3), isoprene, and its main oxidation products, methyl vinyl ketone (MVK), methacrolein (MACR), and isoprene hydroxy hydroperoxide (ISOPOOH). The focus of SAMBBA was primarily on biomass burning emissions, but there were also several flights in areas of the Amazon forest not directly affected by biomass burning, revealing a background with a signature of biomass burning in the chemical composition due to long-range transport of biomass burning tracers from both Africa and the eastern part of Amazonia. We used the [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio and the hydroxyl radical (OH) indirect calculation to assess the oxidative capacity of the Amazon forest atmosphere. We compared the background regions (CO < 150 ppbv), fresh and aged smoke plumes classified according to their photochemical age ([O3] ∕ [CO]), to evaluate the impact of biomass burning emissions on the oxidative capacity of the Amazon forest atmosphere. We observed that biomass burning emissions disturb the isoprene oxidation reactions, especially for fresh plumes ([MVK + MACR + ISOPOOH] ∕ [isoprene] = 7) downwind. The oxidation of isoprene is higher in fresh smoke plumes at lower altitudes (∼ 500 m) than in aged smoke plumes, anticipating near the surface a complex chain of oxidation reactions which may be related to secondary organic aerosol (SOA) formation. We proposed a refinement of the OH calculation based on the sequential reaction model, which considers vertical and horizontal transport for both biomass burning regimes and background environment. Our approach for the [OH] estimation resulted in values on the same order of magnitude of a recent observation in the Amazon rainforest [OH] ≅ 106 (molecules cm−3). During the fresh plume regime, the vertical profile of [OH] and the [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio showed evidence of an increase in the oxidizing power in the transition from planetary boundary layer to cloud layer (1000–1500 m). These high values of [OH] (1.5 × 106 molecules cm−3) and [MVK + MACR + ISOPOOH] ∕ [isoprene] (7.5) indicate a significant change above and inside the cloud decks due to cloud edge effects on photolysis rates, which have a major impact on OH production rates.
- Article
(3601 KB) -
Supplement
(436 KB) - BibTeX
- EndNote





Terrestrial vegetation emits a significant amount of biogenic volatile organic compounds (BVOCs) to the atmosphere, corresponding to 1150 Tg carbon year−1. The most abundant BVOC is isoprene (C5H8), with an annual global emission ranging from 440 to 660 Tg carbon year−1, depending on driving variables such as temperature, solar radiation, leaf area index, and plant functional type (Guenther et al., 2006). In contrast, the global emission rate of anthropogenic, non-methane volatile organic compounds is around 145 Tg carbon year−1 (Janssens-Maenhout et al., 2015). The atmosphere has a natural mechanism to balance the VOCs emitted and their degradation via a complex chain of oxidation reactions, not yet fully understood, followed by the deposition of later-generated products, mostly secondary organic aerosols (SOAs; Prinn, 2014). These oxidation reactions occur mainly through the hydroxyl free radical (OH), which has been often used to express the oxidative capacity of the atmosphere. Therefore, the VOCs play an important role in the atmospheric chemistry, influencing the concentrations of ozone (O3) and OH as well as the conversion rates of nitrogen oxides (NOx= NO + NO2). The VOCs also affect the atmospheric SOAs, which alter the solar radiation budget and cloud droplet nucleation. Moist regions with high availability of solar radiation, such as the Amazon region, affect the VOCs oxidation through the photochemical OH production from O3.
The Amazon is the largest and most diverse rainforest in the world, comprising about 390 billion broadleaf trees of 16 000 distinct species (Ter Steege et al., 2013). The Amazon Basin encompasses about 7 million km2, including territories of Brazil, Bolivia, Peru, Ecuador, Colombia, Venezuela, Guyana, Suriname, and French Guiana, with a significant portion almost untouched by human activity with their natural environmental features preserved. The BVOC mixing ratios in the Amazon are variable, with values ranging from 2.4 to 7.8 ppbv, depending on location, altitude, and seasonal behavior of radiation, temperature, and phenology (Yáñez-Serrano et al., 2015 and references therein). Harley et al. (2004), for example, estimated that about 38 % of the plants in the Amazon forest emit isoprene. Also, studies have shown that the capacity of plants for producing and storing isoprenoids is very specific (Laothawornkitkul et al., 2009; Sharkey et al., 2008).
The atmosphere of the Amazon, in its undisturbed state, oxidizes the BVOCs naturally emitted by the forest vegetation, recycling some OH and depositing reactive carbon back to the surface as several oxidation products, including SOA. In this way, the cleaning process also acts as a local recycling mechanism, preventing the loss of essential nutrients from the forest (Lelieveld et al., 2008). It is estimated that about 90 % of the isoprene and 50 % of the terpenes ((C5H8)n) are removed from the atmosphere via oxidation by OH, followed by the deposition of oxidized VOC and SOA within a timescale of a few hours (Monks, 2005). In fact, isoprene is an important compound in the atmospheric chemistry over forest regions because of its abundance and high reactivity with OH (Barket et al., 2004; Prinn, 2014).
For several years, the traditional understanding was that the unpolluted atmosphere, defined by low levels of nitrogen oxides (NOx), has low concentrations of OH around midday, typically 1–5 × 105 molecules cm−3; however, known discrepancies between atmospheric chemistry model results and observations raised the supposition of a missing OH source (Warneke et al., 2001; Whalley et al., 2012). Recently, airborne measurements performed in an unpolluted atmosphere over the Amazon rainforest found unexpected high oxidative capacity levels, which, complemented with laboratory and numerical modeling studies, led to a different hypothesis for OH production (Lelieveld et al., 2008; Paulot et al., 2009). Concentrations of OH around 5.6 (±1.9) × 106 molecules cm−3 were measured in the planetary boundary layer (PBL) over the Amazon, concomitant with CO, NO, and O3 mixing ratios of 113 (±13.9) ppbv, 0.02 (±0.02) ppbv, and 18.5 (±4.6) ppbv, respectively, values typical of the unpolluted atmosphere. This work pointed to the reaction of isoprene with peroxy radicals (HO2) as an alternative pathway to OH production in an unpolluted environment (Lelieveld et al., 2008). Other OH observation studies conducted in pristine rainforests showing low NO and high isoprene concentrations have consistently reported unaccountably high OH levels, e.g., Whalley et al. (2011). Rohrer et al. (2014) compiled several previous OH observations in environments characterized by large VOC concentrations, such as forested areas, and concluded that it requires a substantial OH recycling mechanism to reconcile the discrepancy between observations and model outcomes based on the conventional understanding of isoprene photo-oxidation (Logan et al., 1981). However, a different school of thought considers these discrepancies between model and observation of OH production due to instrument artifacts. Mao et al. (2012) directly demonstrated the magnitude of potential instrument artifacts by adapting a novel background characterization method called a chemical removal technique, a method to measure OH in parallel with the traditional fluorescence assay with gas expansion (FAGE). The study also illustrated that the application of the chemical removal technique results in agreement between observed and model-calculated diurnal OH variations based on the conventional isoprene photo-oxidation. The same research group also deployed this instrumentation in a rural Alabama forest site as a part of the Southern Oxidant and Aerosol Study (SOAS) campaign (Feiner et al., 2016) and found high isoprene concentrations (up to 10–20 ppb) and low NO levels (∼ 50 ppt) in the afternoon. In this photochemical environment, the observed OH with the chemical removal technique agrees well with the model-calculated OH based on the conventional isoprene photo oxidation scheme. More recently, significant advances have been made with organic peroxy radicals (RO2) produced as intermediates of atmospheric photochemistry, showing the importance of the subsequent reaction pathway to isoprene chemistry (Lew et al., 2018; Teng et al., 2017). The accurate understanding of the isoprene chemistry is required for quantitative predictions of particulate matter concentration, oxidation capacity, and consequent environmental and climate impacts (Liu et al., 2016).
Although Amazonia is mostly dominated by pristine areas, commonly described as a low NO region, there are regions that have been strongly impacted by human activity. The most devastating example is the ongoing deforestation, followed by vegetation burning to open areas for pasture and agriculture production. During the austral winter (from July to October), the Amazonia climate is typically dry and is disturbed each year by extensive vegetation fires in areas of deforestation and agricultural or pasture land management, particularly along the so-called deforestation arc, an area of about 500 000 km2 extending from the southwestern to the eastern border of the forest (Artaxo et al., 2013). During the fire events, an intricate myriad of chemical and physical processes occurs. The continuous increase in temperature of the fresh biomass caused by nearby fires can distill species absorbed by plants with low boiling point (e.g., Tisoprene≅ 307 K), macromolecular bonds can be broken (i.e., low-temperature pyrolysis), gasification reactions converting carbon in the solid char to CO and CO2 can occur and the flames efficiently oxidize the volatile gases to species such as H2O, CO2, and NOx (Bertschi et al., 2003; Longo et al., 2013). The release of isoprene and other BVOCs is dependent on the different phases of biomass combustion, and diverse vegetation communities affect the amount and diversity of VOCs released (Ciccioli et al., 2014). In this disturbed atmosphere, the assumed natural and efficient OH recycling mechanism is affected, altering the oxidative capacity of the atmosphere.
In the absence of biomass burning emissions, isoprene is the dominant reactive VOC in the pristine Amazon forest, and during the day, isoprene oxidation dominates the OH chemistry producing, among other products, methyl vinyl ketone (MVK), methacrolein (MACR), and isoprene hydroxy hydroperoxide (ISOPOOH) (Karl et al., 2007; Liu et al., 2016; Rivera-Rios et al., 2014). In a smoky atmosphere, isoprene oxidation also mainly produces MVK and MACR; however, the molar yields can slightly differ from the ones for the unpolluted condition (i.e., low levels of NO). The updated chemistry of isoprene degradation in the Master Chemical Mechanism (MCM v3.3.1; Jenkin et al., 2015) reported molar yields of about 47 and 34 % for MVK, and 20 and 23 % for MACR, in low (0.1 ppbv) and high (10 000 ppbv) NO level environments, respectively. The calculated yields are consistent with the reported yields studies, although related directly to a specific environment.
In the context of an Amazon rainforest impacted by anthropic and by biogenic emission sources, the airborne measurements conducted in Amazonia during the South AMerican Biomass Burning Analysis (SAMBBA) in 2012 included several fire emission tracers, as well as isoprene and its oxidation products. SAMBBA flights were carried out in both regions directly and indirectly affected by fire emissions. In this work, we analyzed SAMBBA measurements to assess the impact of smoke on the oxidative capacity of the atmosphere in the Amazon region. Due to the lack of direct measurements of OH during SAMBBA, we used the ratio of the mixing ratios of isoprene oxidation products (MVK, MACR, and ISOPOOH) to isoprene as a proxy for the OH levels.
Motivated by the discrepancies between model and observation of OH production in the atmosphere and the influence of the biomass burning plumes in the isoprene reactivity with OH during the day, we propose, in this study, a refinement in the OH estimation method that has been applied by several previous studies (Apel, 2002; Karl et al., 2007; Kuhn et al., 2007; Stroud et al., 2001).
The paper is structured as follows. In Sect. 2, we present the SAMBBA field campaign, including the meteorological conditions and fire occurrence during the campaign period, along with the airborne measurements discussed in this study. The classification method of flight tracks, as well as the method for the indirect OH calculation, are also covered in Sect. 2. In Sect. 3, we presented and discussed the ambient distribution of chemical compounds in the atmosphere (CO, NOx, O3, and isoprene) during SAMBBA, the factors that affected the ratio [MVK + MACR + ISOPOOH] ∕ [isoprene] and the oxidative capacity in distinct environments. Finally, in Sect. 4 the main findings are summarized.
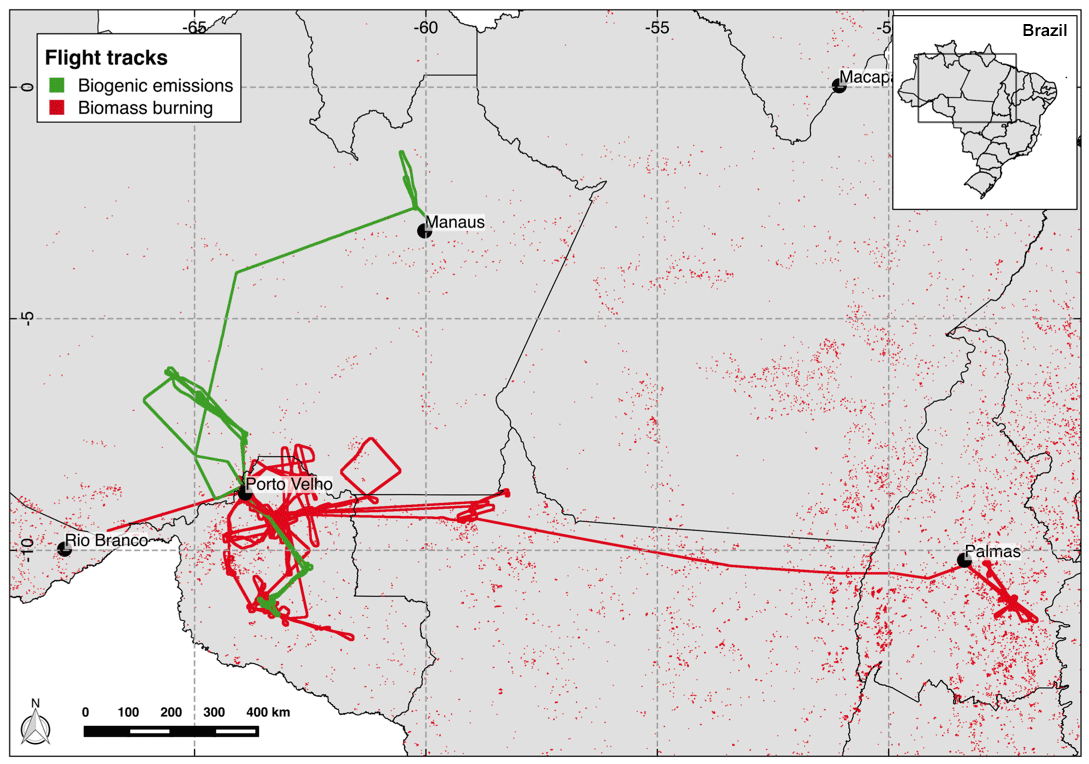
Figure 1SAMBBA flights tracks according to their original goals as biogenic emissions (green) and biomass burning (red). The black dots indicate the locations of the taking-off and landing airports. The red points depict the fires detected by MODIS on board AQUA satellite during the SAMBBA campaign from 14 September to 3 October 2012.
2.1 SAMBBA field campaign
The SAMBBA field campaign was an airborne experiment carried out in the Brazilian Amazonian sector late in the dry season and during the transition from the dry to the wet season, from 14 September to 3 October 2012. Numerous atmospheric measurements were conducted on board the BAe-146 research aircraft, during 20 research flights and 67 flight hours. The BAe-146 research aircraft, from the Facility for Airborne Atmospheric Measurements (FAAM – http://www.faam.ac.uk, last access: 1 June 2018.), was based in Porto Velho – RO, but made use of other regional airports (Palmas – TO, Rio Branco – AC, and Manaus – AM airports) to extend the operational range of the aircraft (Fig. 1). During SAMBBA, the areas with positive anomalies of precipitation were mostly in western and central Amazonia, while the eastern sector was drier than the climatic average. The mean daily rainfall east of the SAMBBA flight area was typically below 1 mm. In contrast, in the western and central part, the mean daily precipitation ranged from 3 to 10 mm because of an intense cold front incursion, an early precursor of the dry-to-wet transition season. As a result, the fires in the western part of Amazonia, where most SAMBBA flights took place, were scattered and intermittent. The most intense and persistent fire activity occurred in the eastern part. The aerosol optical depth (AOD) in Porto Velho dropped from the typical 1.5 (channel 550 nm) in the first half of September 2012 to below 0.5 during SAMBBA; in contrast, the AOD was constantly above 1 in the eastern part of Amazonia.
Table 1SAMBBA research flights analyzed in this work. Reference locations indicated in the map of the Fig. 2.
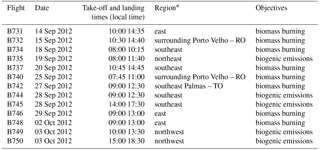
*The “Region” column lists the directions from Porto Velho – RO.
The BAe-146 research aircraft flew with a comprehensive suite of instrumentation, measuring aerosols and cloud microphysics properties, chemical tracers, radiative fluxes, and several meteorological variables. Essential for this work were measurements of isoprene, MVK, MACR, and ISOPOOH, which were carried out using an onboard proton transfer reaction mass spectrometer (PTR-MS, Ionicon, Innsbruck, Austria) with a quadrupole detector and a typical cycle time around 3–5 s. The instrumental, operational, and calibration details are described in Murphy et al. (2010), but it is pertinent to note that (1) the quadrupole detector cannot distinguish between the isobaric molecules MVK and MACR, and its decomposition interferer ISOPOOH, so it reports the data at m∕z 71 as the sum of three isomers, even though it was only calibrated for MVK + MACR (Liu et al., 2016); (2) the conversion yields of ISOPOOH into MVK and MACR was observed to be greater than 70 %, but the decomposition is known to be highly sensitive to instrumental settings such as temperature, contact time, and type of surface materials, especially transition metal surfaces (Liu et al., 2013; Nguyen et al., 2014; Rivera-Rios et al., 2014, Liu et al., 2016, Bernhammer et al., 2017); and (3) there is a well-known interference in the isoprene signal at m∕z 69 in biomass burning plumes from furan. The PTR-MS was calibrated post-flight using a calibrated gas standard provided by Apel Riemer Environmental Inc. We compared the PTR-MS isoprene data with the isoprene data derived from the whole air sampling (WAS) system on the aircraft to correct the PTR-MS isoprene data due to a probable interference from furan at m∕z 69 in biomass burning plumes. The WAS system was described in Hopkins et al. (2011) and consists of discrete air samples collected in 3-liter silco-treated stainless steel canisters with subsequent post-flight analysis by GC-FID. In the background environment, the agreement between the two systems was excellent (isoprenewas ∕ isopreneptrms= 0.81, SD = 0.56), while in biomass burning regions we estimated a high furan contribution in fresh (isoprenewas ∕ isopreneptrms= 0.25, SD = 0.12) and aged (isoprenewas ∕ isopreneptrms= 0.77, SD = 0.57) smoke plumes. The isoprene data have been adjusted accordingly.
In addition, NO measurements were conducted using a chemiluminescence instrument (Air Quality Design Inc., Wheat Ridge, CO, USA), with the NO2 measured using a second channel after photolytic conversion to NO. The photolytic conversion eliminates the possible interference from NOz on the NO2 channel. The detection limits were close to 10 pptv for NO and 15 pptv for NO2 for 10 s averaged data, with estimated accuracies of 15 % for NO at 0.1 ppbv and 20 % for NO2 at 0.1 ppbv (Allan et al., 2014). For the O3 and CO analyses, we used the TEi49C and AL5002 VUV fast fluorescence onboard instruments, respectively (Gerbig et al., 1996, 1999; Palmer et al., 2013). Calibration gases were supplied to the rack from the gas bottle stowage, and the air sampling from the atmosphere was via the air sample pipes and a dedicated window-mounted inlet system.
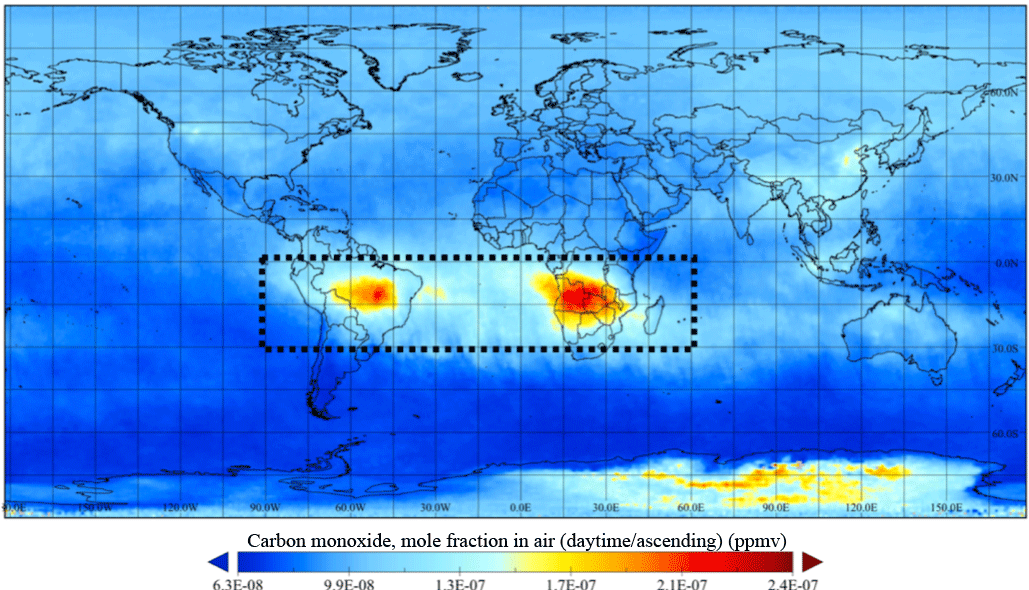
Figure 2Time-averaged CO (ppmv) during the SAMBBA period (14 September–3 October 2012) from AIRS on board the AQUA satellite during daytime at 500 hPa. The region of interest is indicated on the map.
2.2 Classification method of flight tracks
During the planning phase, SAMBBA flights were classified according to their scientific objectives as either biogenic or biomass burning flights (Fig. 1). For this study, we selected 13 flights according to the gaseous chemistry data available (Table 1). Additionally, we only considered the data collected below 2000 m and between 11:00 and 18:00 LT (local time) to capture the difference in the oxidative capacity along the altitude during daytime, since the OH concentration is regulated by photochemistry (Elshorbany et al., 2009). Despite the classification in the planning phase, parts of some flight tracks passed through unpolluted regions, smoke haze, or even interception of fresh smoke plumes. To maximize the use of data, we classified parts of the flight tracks according to the CO mixing ratio values as background (BG) and biomass burning. According to Andreae et al. (2012) and several references therein, the Amazon rainforest atmosphere has a background CO mixing ratio typically around 100 ppbv. However, the mean CO inflow into the Amazon Basin during the SAMBBA period at 500 hPa, retrieved from Atmospheric Infrared Sounder (AIRS) measurements on board the AQUA satellite, ranged between 140 and 160 ppbv (Fig. 2 and Supplement Fig. S7). This hemispheric inflow is homogeneous along the vertical column up to around 400 hPa. In fact, there were only few SAMBBA samples with CO mixing ratio values below 100 ppbv. Therefore, we adopted a threshold of 150 ppbv to represent the background of CO in the Amazon atmosphere during the SAMBBA campaign.
Table 2Observations of the enhancement ratio and plume age in tropical and subtropical sites.
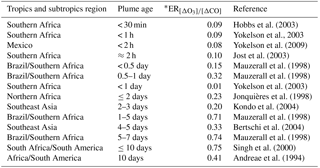
∗ The single value for enhancement ratio presented here represents the mean measurement.
As O3 is formed photochemically downwind during smoke aging, the enhancement ratio of O3 to CO is acceptable as a reliable indicator of the smoke plume age (Andreae et al., 1994; Parrish et al., 1993). Furthermore, due to the lack of NOy ∕ NOx ratio in SAMBBA, we used the ratio of O3 to CO as a proxy for smoke plume age. The biomass burning flight tracks with [CO] > 150 ppbv were then reclassified as fresh smoke plume (FP) or as aged smoke plume (AP) interceptions according to the following.
During the BG flight tracks (CO ≤ 150 ppbv), the mean value of the O3 mixing ratios near the surface (< 500 m) was 21 ± 7 ppbv, which we then adopted as the O3 mixing ratio background. In Table 2, we list the values of and the estimated smoke plume age for several smoke measurements in Amazonia and Africa. Jost et al. (2003) found the value of 0.1, 2 h after emission in Otavi, northern Namibia; and Andreae et al. (1988) found 0.08 for fresh biomass burning (650 m altitude) in the Amazon Basin region. Comparable values of (0.09) were observed in other young smoke plumes with 0.5–1.0 h aged during the Southern African Regional Science Initiative 2000 – SAFARI 2000 (Hobbs et al., 2003; Yokelson et al., 2003). Mauzerall et al. (1998) reported 0.15 for FPs with fewer than 4.8 h over regions with active fires in the northeast region of Brazil and in Africa. In short, we classified the parts of the flight tracks as background (BG) when [CO] ≤ 150 ppbv, and the biomass burning flights ([CO] > 150 ppbv) into two subgroups of FP, with [O3] ∕ [CO] < 0.1 and AP, with [O3] ∕ [CO] ≥ 0.1.
2.3 Method description for the OH calculation
The OH concentrations from the [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio using the sequential reaction model were originally developed by Apel (2002) and Stroud et al. (2001) and modified according to the approach of Karl et al. (2007). This method can be used to investigate the impact of vertical transport, representing the processing time of the isoprene and its oxidation products from the surface to the atmosphere through the ratio of PBL depth and the convective velocity scale. To have a more accurate OH estimation, we modified the processing time t to represent not only the vertical transport but also the horizontal atmospheric circulation, where t was calculated as a function of the enhancement ratio (see Supplement Fig. S5). Table 2 shows the plume age time and the enhancement ratio values used in the new approach. This method is based on observations that the isoprene reaction rate with OH (rate coefficient ≅ 1.0 × 10−10, lifetime ≅ 1.4 h) is more important than with O3 (rate coefficient ≅ 1.3 × 10−17, lifetime ≅ 1.3 day) during the daytime and assuming a constant reaction rate. Following the simplified sequential reaction model, we can estimate OH concentration, in molecules cm−3, with the following analytical expression:
where kiso and kprod are, respectively, the reaction rate constants of isoprene + OH (1.1 × 10−10 cm3 molecules−1 s−1), and [MVK + MACR + ISOPOOH] + OH (6.1 × 10−11 cm3 molecules−1 s−1), and we are assuming a total yield of 0.55 of MVK + MACR + ISOPOOH from the OH + isoprene reaction (Apel, 2002; Karl et al., 2007). We estimated the processing time as (seconds), which is the fitting function of several previous measurements of (Supplement Fig. S3) and plume age observations in tropical and subtropical sites (Table 2).
Table 3Airborne measurements of CO, NOx, and O3 mixing ratios in Amazonia and cerrado areas in Brazil.
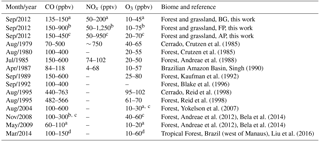
Measurements in a background, b fresh plumes, and c aged plumes. d Measurements with a background and maximum interval. The data without an index was collected without any particular criteria.
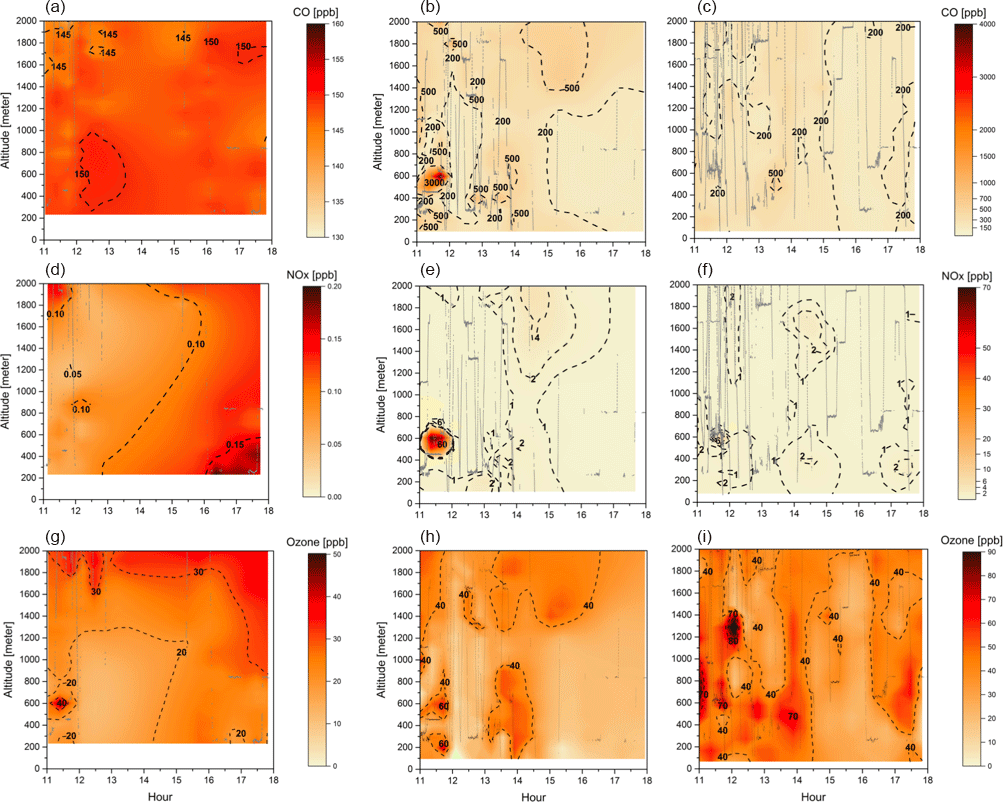
Figure 3Cross section of CO (a–c), NOx (d–f), and O3 (g–i) mixing ratios (ppbv) for the three different groups: background environment (on the left), fresh smoke plume (t < 2 h, in the middle), and aged smoke plume (t > 2 h, on the right). The aircraft data were interpolated from the various vertical profile measurements using the kriging correlation method (Supplement Fig. S1). Grey lines show the flight tracks. Hour units are presented in local time.
3.1 Ambient distributions of CO, NOx, and O3
Figure 3 depicts CO, NOx, and O3 mixing ratios measured at different altitudes, up to 2 km, and time of the day, between 11:00 and 18:00 LT. In Fig. 3, the flight tracks are separated according to the BG, FP, and AP classification, while Table 3 shows typical values of CO, NOx, and O3 mixing ratios measured in this study and during several previous airborne campaigns in Amazonia and savannah areas in Brazil. During the SAMBBA field experiment, in BG conditions (i.e., CO < 150 ppbv), the NOx mixing ratio ranged from 50 to 200 pptv. Torres and Buchan (1988) reported measurements of NO mixing ratios ranging between 20 and 35 pptv during the Amazon Boundary Layer Experiment (ABLE-2A) between July and August 1985. Modeling results of Jacob and Wofsy (1988) found NOx mixing ratio values around 200 pptv, with the NO mixing ratio values similar to the ABLE-2A observations that were conducted over the Amazon rainforest. Also in the Brazilian Amazon Basin during the wet season, aircraft measurements as part of the NASA Amazon Boundary Layer Experiment (ABLE-2B), showed NOx mixing ratios ranging from 4–68 pptv (Singh, 1990). Comparing our results with these previous studies, the SAMBBA experiment showed a slight influence from polluted regions. More recently, Liu et al. (2016) used four sets of different MCMs and estimated that NO mixing ratio using the NO vs. HO2 isoprene chemistry (fHO2 : fNO ∼ 0.6–1.4) would be around 20–40 pptv of NO based on measurements in the Amazon, lower than that obtained in our study. On the other hand, flight tracks in biomass burning areas showed high values of NOx in FP (50–1250 pptv) and AP (50–950 pptv) compared with other studies in forest areas of Amazonia, including a study in the cerrado area in Brazil (∼ 750 pptv) conducted by Crutzen et al. (1985) during the dry season.
The O3 mixing ratios in the BG environment reached 40 ppbv at about 600 m altitude during flight B735 (at 11:30 LT, Fig. 3), although typical values ranged from 10 to 45 ppbv (< 2000 m; Table 3). This value of 40 ppbv at 600 m altitude is nearly 2 times the mean value of the O3 mixing ratio that we used as background in the enhancement ratio, and even out of the range of the standard deviation (21 ppbv, SD = 7). As a secondary pollutant, O3 is commonly found in low concentrations near the surface, a fact not observed in our study even for the BG samples. Bela et al. (2015) reported O3 ranging from 10 to 20 ppbv during the wet-to-dry transition season (May–June 2009), measurements performed in a clean atmosphere during the Regional Carbon Balance in Amazonia (BARCA-B) campaign. In contrast, during the BARCA-A campaign in November–December 2008 (during the dry-to-wet transition season), the O3 mixing ratio ranged from 40 to 60 ppbv in an area influenced by fires, values which are comparable with FP (10–75 ppbv) and AP (20–70 ppbv), and even in BG conditions (10–45 ppbv) during SAMBBA.
In terms of CO and NOx, flight tracks classified as FP were the most polluted of the campaign. The CO and NOx mixing ratios for FP reached, respectively, values above 3000 and 60 ppbv at 600 m from the surface between 11:00 and 12:00 LT. The enhancement of CO and NOx mixing ratios near the surface suggests significant vertical transport due to the hot plume buoyancy, which may increase the tracer lifetime released to the atmosphere. The vertical transport can be observed mainly for CO at different altitudes and time of day, since the CO is preserved longer along the plume when compared with NOx. The measurements in fresh biomass burning plumes also capture higher levels of CO mixing ratios (≅ 500 ppbv) at 1.4 and 2 km of altitude. Yokelson et al. (2007), during the Tropical Forest and Fire Emissions Experiment (TROFFEE), reported a vertical transport mechanism called a “mega-plume” at 2 km altitude during a flight south of the Amazon rainforest (from Manaus to Cuiabá), with the CO mixing ratio reaching 1200 ppbv. The TROFFEE experiment used airborne measurements during the 2004 Amazon dry season, and reported, on 8 September, the presence of a massive plume formed by numerous fires. During SAMBBA campaign, we also detected the presence of similar plumes during flight B742, classified mostly as FP, with a unique CO mixing ratio value, peaking at ∼ 5000 ppbv at about 600 m. These results demonstrate the strength of vertical transport during a fresh biomass burning event, with the plume injection height up to 2 km. Freitas et al. (2006, 2007, 2010) highlighted the importance of representing the injection height of biomass burning plumes in numerical models to describe the regional smoke distribution. Trentmann and Andreae (2003) also demonstrated a large impact of fire emissions on the chemical composition in a young biomass burning plume using a 3-D chemical transport model and direct observations. These authors reported simulated high values near the fire (z ≅ 150 m) for CO and NOx, with mean values around 18 000 ppbv and 404 ppbv, respectively. On the other hand, Andreae et al. (2012) reported CO mixing ratios up to 400 ppbv in a smoky region in the southern Amazon Basin during the BARCA-A experiment. The highest values were found at about 1000 m altitude, late in the dry season (November 2008). In this study, the CO mixing ratio measurements in FP and AP ranged from 150 to 900 ppbv and from 150 to 450 ppbv, respectively. The CO mixing ratios in FP (150–900 ppbv) are comparable with values found by Reid et al. (1998) in a cerrado area (440–763 ppbv) and some forest studies (up to 600 ppbv) conducted by Andreae et al. (1988), Kaufman et al. (1992), and Yokelson et al. (2007). The values found in AP agreed with values of CO mixing ratios from forest areas, impacted by smoke haze plumes (Table 3).
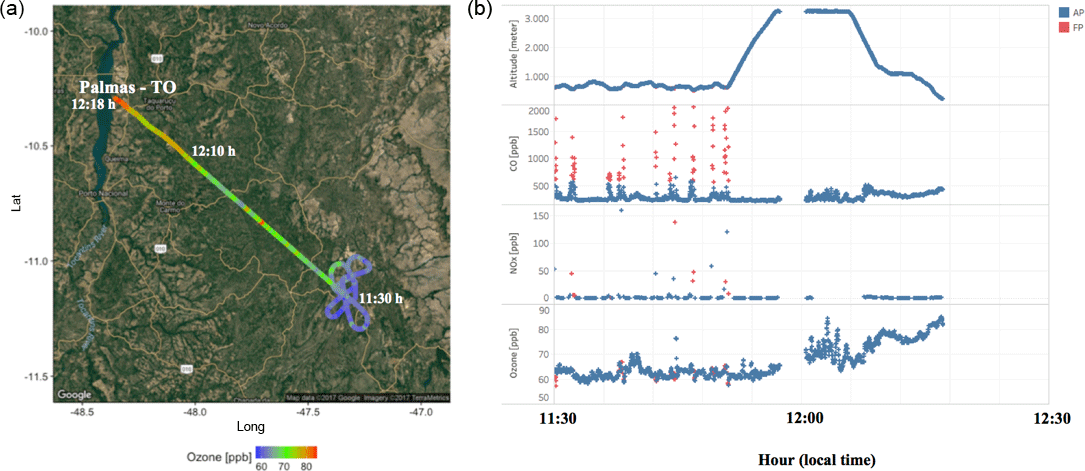
Figure 4(a) the track of flight B742 that landed in Palmas – TO. The color bar represents the measured O3 mixing ratios (ppbv) along the flight track. (b) From top to bottom, the altitude and the CO, NOx, and O3 mixing ratios (ppbv) measured along the B742 flight track. The red and blue dots represent the parts of the flight track classified as fresh (FP) and aged (AP) smoke plumes, respectively.
In Fig. 3, during most of the flight tracks classified as AP, the NOx mixing ratio values were below 2 ppbv, except for a peak of 6 ppbv at 600 m from the surface between 11:30 and 12:00 LT, a value below that observed in the FP environment (60 ppbv). Conversely, the O3 mixing ratio results in FP presented high levels (≅ 80 ppbv) around 12:10 LT at 1300 m altitude. We also investigated the high levels of CO and NOx mixing ratios found in FP corresponding to the same flight in which we also detected high levels of O3 in AP. In fact, Fig. 4 shows the track of flight B742 in the transition from a remote site impacted by FP to an urban site impacted by AP in Palmas – TO (O3 ≅ 80 ppbv). The mean value of O3 found in FP was 31 ppbv (SD = 14), which is 29 % lower than measured for AP (44 ppbv, SD = 13 ppbv), since O3 is produced as a secondary product from the interaction between VOCs and NOx. The O3 mixing ratios in FP peak at about 60 ppbv near the surface (200–600 m) and, for most cases, plumes with about 40 ppbv were observed both near the surface and in high altitudes (Fig. 3). During the TROFFEE experiment, Yokelson et al. (2007) found O3 mixing ratios of about 30 ppbv in smoke haze layers in Amazonia. Our results showed higher values for O3 mixing ratios, ranging from 10 to 75 ppbv in FP and 20 to 70 ppbv in AP. Agreeing with SAMBBA results, Reid et al. (1998) found O3 mixing ratios ranging from 60 to 100 ppbv during the SCAR-B experiment in the 1995 dry season, and Kaufman et al. (1992) found similar levels of O3 in a forest site in Amazonia during BASE A (Table 3).
Table 4Airborne measurements of isoprene, oxidation ratio [MVK + MACR + ISOPOOH] ∕ [isoprene], and OH in remote areas and biomass burning environments worldwide. Standard error reported in parentheses for this study.
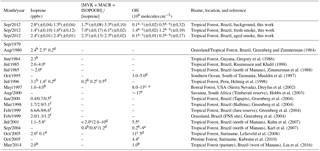
a Estimated values. Measurements at b surface, c boundary layer, d free troposphere, and e cloud layer. f Measurements at 9–12 h/12–15 h/15–18 h.

Figure 5Cross section of the isoprene mixing ratio (ppbv) (a–c) and the [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio (d–f) for the three different groups: background environment (on the left), fresh smoke plume (t < 2 h, in the middle), and aged smoke plume (t > 2 h, on the right). The aircraft data were interpolated from the various vertical profile measurements using the kriging correlation method (Supplement Fig. S1). White dashed lines show the flight tracks. Hour units are presented in local time.
3.2 Isoprene and its oxidation ratio
Information about isoprene transport and chemistry can be derived from the isoprene abundance in the atmosphere and the ratio of its oxidation products over isoprene, [MVK + MACR + ISOPOOH] ∕ [isoprene]. During the day, the isoprene chemistry is mainly affected by the distance from the emission source (transport time), photochemical degradation, and availability of OH, which react with isoprene to produce (among other chemical species) MVK, MACR, and ISOPOOH (Kuhn et al., 2007; Liu et al., 2016). During SAMBBA, the mean isoprene mixing ratio in BG was 2.8 ppbv and 1.5 ppbv for the boundary layer and cloud layer, respectively. We also detected higher values of O3 (40 ppbv at 600 m) in BG, shown in Fig. 3, coinciding with the interpolated cross section of isoprene (≤ 4 ppbv at 600 m), also in the BG environment (Fig. 5). As mentioned by Barket et al. (2004), a sequence of reactions initialized by the reaction of isoprene with OH leads to the production of organic peroxy radicals (RO2), which then react with NOx promoting the O3 formation observed during the BG flights tracks. Table 4 summarizes the mean values of isoprene and the oxidation ratio [MVK + MACR + ISOPOOH] ∕ [isoprene] during SAMBBA, and previously reported airborne measurements in remote areas and biomass burning environments. The isoprene mixing ratios measured during SAMBBA in the BG environment agree with values reported in pristine areas of the Amazon forest (Greenberg et al., 2004; Greenberg and Zimmerman, 1984; Gregory et al., 1986; Helmig et al., 1998; Kuhn et al., 2007; Lelieveld et al., 2008; Rasmussen and Khalil, 1988; Zimmerman et al., 1988). Some studies conducted in the Amazonian tropical forest (e.g., Greenberg et al., 2004 and Kuhn et al., 2007) reported isoprene mixing ratios up to ∼ 7 ppbv, values higher than we found during the SAMBBA campaign.
On average, we found a reduction of isoprene in FP (1.4 ppbv), and a more discrete reduction in AP (2.4 ppbv), relative to the BG value (2.8 ppbv), within the PBL (< 1200 m), producing a lower value around 50 % and 14 %, respectively. In contrast, we observed above the PBL (> 1200 m) an impressive increase of about 60 % of the isoprene in AP (2.4 ppbv) relative to the BG value (1.5 ppbv), which is about the same mean value found in FP (1.6 ppbv). These high levels of isoprene at higher altitudes in air masses affected by biomass burning emissions are likely to be associated with the heat released from vegetation fires affecting nearby plants with enough energy to release significant amounts of isoprene to the atmosphere, especially in tropical forest fires in Brazil (Ciccioli et al., 2014). Müller et al. (2016), for example, found isoprene mixing ratios up to 15 ppbv in a smoke plume from a small forest fire in Georgia, USA. We also found higher isoprene mixing ratios in the upper levels (> 1200 m) of smoke areas when compared with pristine mixed layer studies mentioned previously. These results reinforce the hypothesis that fire activity promoted the isoprene transport to higher altitudes both in fresh (≅ 6 ppbv, 1700–2000 m) and aged plumes (≅ 4 ppbv, 1600–2000 m) during SAMBBA flights (Fig. 5). In Fig. 6, we also verified the average isoprene mixing ratio (< 2000 m) in AP (2.4 ppbv) was 71 % higher than FP (1.4 ppbv) and similar to the mean value measured in BG (2.6 ppbv).
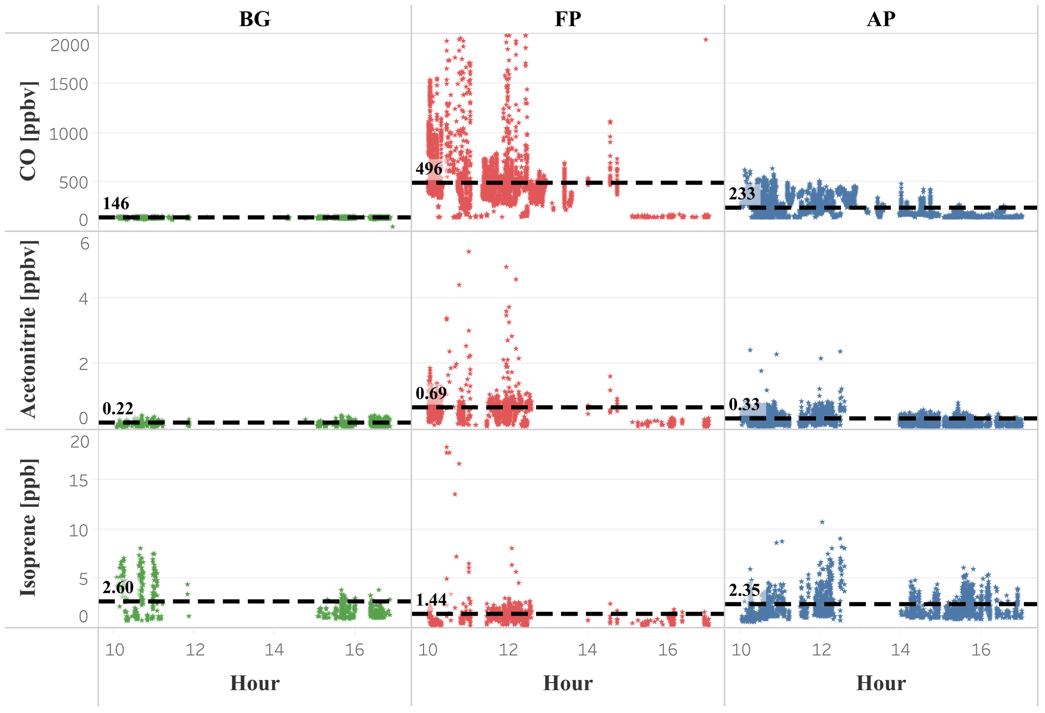
Figure 6Isoprene, acetonitrile, and CO mixing ratios (ppbv) as a function of daytime (local time) for the different chemical regimes previously classified as background (green dots), fresh smoke plume (red dots), and aged smoke plume (blue dots). Black dashed lines, and the numbers next to them, represent the mean values of the measurements taken below 2000 m altitude. Hour units are presented in local time (11:00–18:00 LT).
In smoke plumes, biomass burning tracers, such as acetonitrile and acetaldehyde, are present at high concentration, while the [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio is low. In contrast, as an aging effect, smoke plumes typically have higher values for the [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio, since there is more time for the isoprene degradation. According to Apel et al. (2002), the high value for kOH is responsible for the majority of the chemical processing of isoprene by OH. As the rate constant of OH with MVK and MACR are lower than isoprene-OH, we expect an increase in the ratio [MVK + MACR + ISOPOOH] ∕ [isoprene], especially in a polluted environment in the boundary layer. Figure 7 presents the plume interception during the flight B732, between 10:00 and 11:30 LT, in which it is possible to observe the different altitude interceptions through the biomass burning tracers and [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio. In this study, we found the [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio mean value ranging from around 1.7 in the boundary layer up to 3.3 in the cloud layer for BG conditions, with AP presenting a similar value (2.3) for both boundary layer and cloud layer. In contrast, FP had the highest value in the boundary layer (7.0) and cloud layer (6.1), values reported by Kuhn et al. (2007), in the tropical forest in Brazil (north of Manaus).
We did not find any substantial variation above the PBL in [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio associated with the presence of smoke in AP (2.8) but did find an increase in the BG value (3.3) in the upper levels (> 1200 m). The FP is more active within the boundary layer than in upper levels, with the isoprene oxidation ratio about 6.1 above 1200 m. Comparable with our results in FP, Kuhn et al. (2007) during the Cooperative LBA Airborne Regional Experiment (LBA-CLAIRE-2001), also found [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio values up to 2, below 1000 m of altitude, and between 2 and 10, within the 1000–2000 m vertical layer. In summary, we found a strong increase in the isoprene oxidation ratio from the surface up to 2000 m for FP relative to the BG and AP observed during SAMBBA and other previous studies in biomass burning environments (Table 4). The energetic process that occurs during the biomass burning causes the isoprene plume to be transported rapidly to higher levels, impacting the isoprene oxidation level, with FP samples presenting a higher [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio.
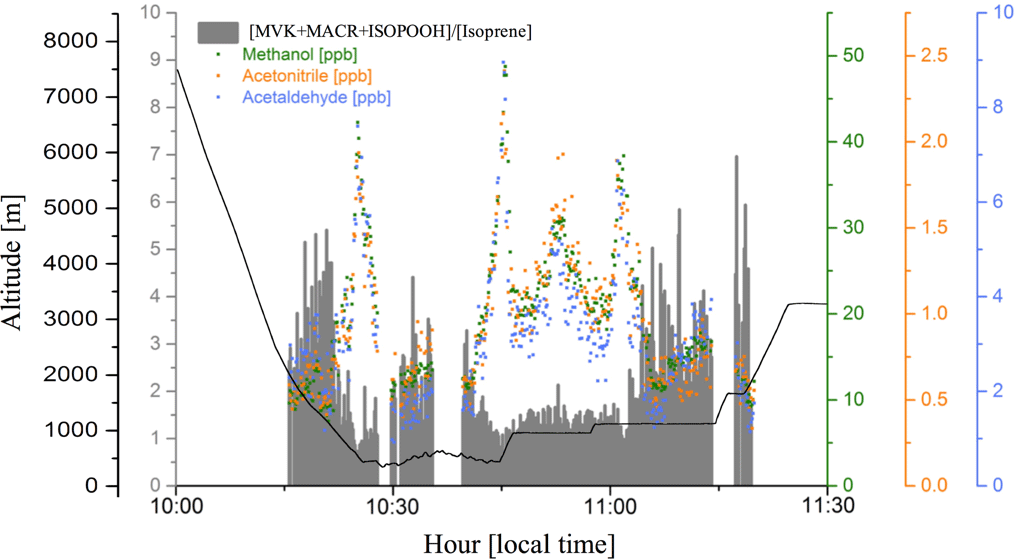
Figure 7Methanol (green dots), acetonitrile (orange dots), and acetaldehyde (blue dots) mixing ratios (ppbv), and the [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio (gray bars), during a plume interception along the flight track B732 in different altitudes.
We also observed during SAMBBA campaign values of the [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio above 6 in BG air masses above 1800 m and between 12:00 and 13:30 LT. In contrast, in FP and AP, values in the range 4–6 are equally distributed in the vertical profile, with some high values near the surface (Fig. 5). Along the cloud layer (1200–2000 m), we found that isoprene oxidation in BG environment increase (94 %) as in FP levels (Table 4). Karl et al. (2007) also reported evidence of an increase in the oxidizing power of the atmosphere in the transition from PBL to cloud layer (1200–1900 m) during the TROFFEE experiment, with the [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio ranging from 0.39 up to 1.2 between 300 and 1800 m, already into the cloud layer (CL). Although lower values for the [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio were found during TROFFEE compared with LBA-CLAIRE, both studies have suggested the occurrence of an oxidizing power in the transition from the PBL to the CL. In both cases, there was a positive gradient, increasing the [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio. Furthermore, Helmig et al. (1998) reported similar behavior in a remote Peruvian Amazonia site, with the [MVK] ∕ [isoprene] ratio equal to 0.15, 0.19, and 0.48, near the surface (≅ 2 m), in the PBL (91–1167 m), and above the PBL (1481–1554 m), respectively. The [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio increasing toward the top of the PBL and CL is likely to be due to the enhancement of the photolysis rates. Direct experimental data reported by Mauldin et al. (1997) also indicated significant changes above and inside cloud decks due to cloud edge effects on photolysis rates that have a major impact on OH production rates. Figure 8 presents the density distribution for the [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio along several altitude layers during SAMBBA. In FP, the average [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio was 6.3 at 500–1000 m, 7.6 at 1000–1500 m, and returning to 5.9 at 1500–2000 m. These results are consistent with the increase in the oxidative capacity in the transition from the PBL to the CL, reported by Mauldin et al. (1997) and Karl et al. (2007), with SAMBBA measurements showing an average [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio constantly increasing in BG and the highest value at 500–1000 m in AP. The results show that the isoprene oxidation reaction is also enhanced at higher altitudes in the BG environment, increasing from 1.4 at the first 500 m to 3.4 at 2000 m. Another characteristic observed during biomass burning events is their capacity to disturb the isoprene oxidation reactions, especially in the fresh plumes. As showed in Fig. 8, the isoprene oxidation is higher in FPs at lower altitudes (∼ 500) than in aged smoke, anticipating near the surface a complex chain of oxidation reactions which may be related to SOA formation. Rohrer et al. (2014) compared observations of OH radicals in different environments characterized by high VOC concentrations; they found that VOC degradation not only accelerates but also occurs at the maximum rate if NOx is present in adequate amounts. Thus, biomass burning is a source of NOx, favoring an increase in the oxidative capacity. According to Rohrer et al. (2014), the OH recycling mechanism is shown to be active not only in pristine biogenic air masses but also in the interface region between anthropogenic and biogenic emissions, such as in the region surrounding Manaus – AM, where urban and biogenic emissions are mixed.
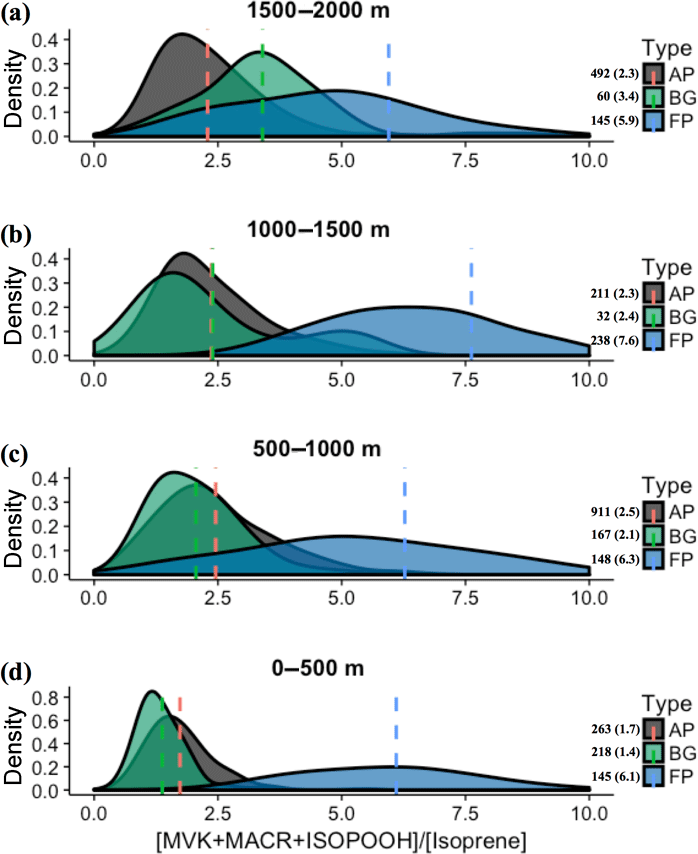
Figure 8Density distributions of the ratio [MVK + MACR + ISOPOOH] ∕ [isoprene], at the altitude layers (a) 1500–2000 m, (b) 1000–1500 m, (c) 500–1000 m, and (d) 0–500 m. The kernel analysis was carried out considering the classification for background (BG), aged smoke (AP), and fresh smoke plumes (FP). The number of samples and mean values for each group are depicted near the color bars.
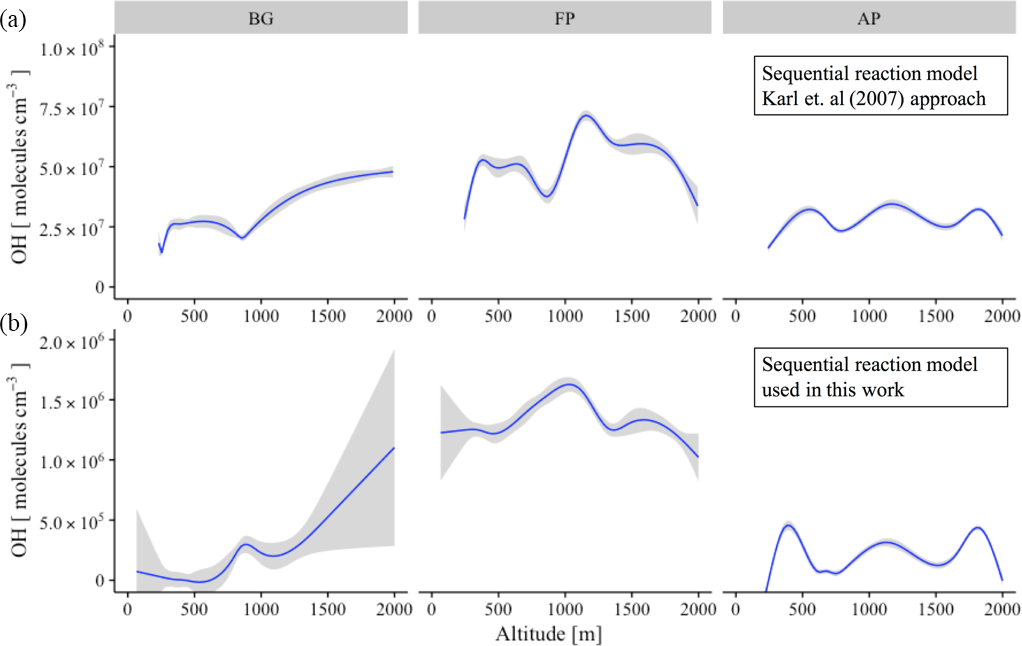
Figure 9Vertical profile of OH concentration (molecules cm−3) for the different chemical regimes: background environment (BG), fresh smoke plume (FP), and aged smoke plume (AP). (a) The sequential reaction model according to the original approach of Karl et al. (2007), and (b), the new approach used in this work. Blue lines are the trend lines and gray intervals represents the level of confidence (0.95) used.
3.3 OH predicted using a sequential reaction approach
The abundance of OH in the atmosphere is determined by equating the kinetic rate of its production and loss. Due to the absence of OH measurements during SAMBBA, we inferred the OH concentrations using a sequential reaction model to the observed profiles of the [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio. Table 4 presents the average values of OH concentration in remote areas and biomass burning environments worldwide, and the estimated OH concentration calculated via the sequential reaction model approaches from Karl et al. (2007) and via the approach proposed in this study. In FP, the estimated OH concentration reached the highest value within the PBL (1.4 × 106 molecules cm−3), when compared with AP or even with the BG environment, both with OH concentration ∼ 0.1 × 106 molecules cm−3. The photochemical environment in young biomass burning plumes differs from the clean conditions, especially near the surface. Hobbs et al. (2003) found OH concentrations of about 1 × 107 molecules cm−3 for a fresh plume from savanna fire in South Africa, values higher than those found in our estimation. In the CL, the estimated OH in AP showed a reduction of 15 % relative to BG (0.5 × 106 molecules cm−3) environment, in opposition to increased pattern in FP (1.2 × 106 molecules cm−3). The estimated OH concentration corroborates the hypothesis that the biomass burning event can intensify the oxidative capacity at low altitudes.
Figure 9 shows the vertical profile of estimated OH concentration in different chemical regimes, comparing the sequential reaction model according to the original approach of Karl et al. (2007) with the new approach used in this work. Throughout the altitude range 0–2000 m, the difference in OH calculation between the two methods was approximately 2 orders of magnitude, although presented a similar pattern in the different chemical regimes. Flight tracks classified as BG tend to increase the OH concentration along the altitude, with the inflection point occurring before the 1000 m. Differing from BG environments, FP presents a decreasing pattern for OH concentration after 1000 m of altitude, with the AP in an intermediate state. Our results suggest that in the FPs, the vertical transport predominates with the oxidative capacity reaching its maximum at 1000 m. In the flight tracks classified as BG, we observed the widest variation in the average OH concentration using the new sequential reaction model (Fig. 9, on bottom), especially in upper levels (0.5–1 × 106 molecules cm−3), although reported a lower confidence level in this region due to a reduced number of samples. In all three different chemical regimes, the vertical profile of OH concentration presented an increase near to CL (∼ 1000 m), in agreement with previous studies (Karl et al., 2007; Kuhn et al., 2007; Langford et al., 2005; Mauldin et al., 1997).
Similar to the OH vertical profile, the amount of NOx increases in the boundary-cloud layer (500–1000 m) for FP and AP, with the O3 pattern presenting ∼ 20 ppbv higher than the values from BG environment. In contrast, the highest values for OH in BG environment was found close to 2000 m of altitude, which reinforce the Lelieveld hypotheses (Lelieveld et al., 2008) of the reaction of isoprene-derived peroxy radicals with organic peroxy radicals as an alternative pathway to OH production in an unpolluted environment.
The estimated OH concentration values presented in this study agree in order of magnitude with most modeled and observed values previously reported for Amazonia and other forest areas. Prediction studies in a forest site at Surinam, conducted by Warneke et al. (2001), estimated a concentration of OH ranging from 1 to 3 × 105 molecules cm−3 (24 h average), and Williams et al. (2001) calculated a range of 0.6–1.1 × 106 molecules cm−3 during daytime. During the Guyanas Atmosphere–Biosphere exchange and Radicals Intensive Experiment with a Learjet (GABRIEL) experiment in October 2005, the observed average OH concentration in the boundary layer (< 1 km) over the Suriname rainforest in the afternoon was 4.4 × 106 molecules cm−3 (Kubistin et al., 2010). On the other hand, Dreyfus et al. (2002) reported high levels of OH concentration (8–13 × 106 molecules cm−3) in the boundary layer over a forest area in Sierra Nevada, California; this forest site was influenced by wind flow patterns, transporting anthropogenic volatile organic compounds and NOx from the Sacramento region toward the Sierra Nevada.
Several studies investigated the uncertainties in the isoprene oxidation mechanism, and most of them focus on OH concentration levels through observational and modeling studies (de Gouw et al., 2006; Kubistin et al., 2010; Kuhn et al., 2007; Lelieveld et al., 2008; Lu et al., 2012; Whalley et al., 2012; Yokelson et al., 2007). Under a high isoprene and low NO atmospheric regime, there is a controversial discussion about the impact on the oxidative capacity in forest sites. Some observations indicate that high OH levels cannot be accounted for by the conventional OH production and recycling mechanisms (Rohrer et al., 2014), but some suggested that the enhanced OH signal is caused by instrumental artifacts rather than the ambient OH (Mao et al., 2012). Our calculated OH with the observational constraints is consistent with previous empirical estimates by Warneke et al. (2001) that estimated OH concentration around 1–3 × 105 molecules cm−3 (24 h average); Williams et al. (2001) also found 0.6–1.1 × 106 molecules cm−3 during daytime without augmented OH recycling mechanisms developed to account for the recent higher than expected OH observations. According to the most recent results in the Amazon rainforest (Liu et al., 2016), the order of magnitude of the OH concentration estimated in our study agrees well with both OH concentrations close to 1 × 106 molecules cm−3.
We present a concise chemical characterization of the atmosphere of Brazilian Amazonia during the SAMBBA airborne experiment from 14 September to 3 October 2012, comprising the transition period from the dry to wet season. SAMBBA flights were carried out in remote areas, as well as areas under the influence of biomass burning that commonly occurs in the region. The flight classification method adopted in this study prioritized the chemical regimes using CO mixing ratios and the enhancement ratio of O3 to CO to categorize different flight tracks to include BG flights, as well as FP and AP flights. In this study, we modified a method to estimate OH concentration values using the sequential reaction model described in Sect. 2.3 for both biomass burning regimes and background environment. Through this new method, and we look for a better estimate of OH in the atmosphere and in the future, we expect to apply the indirect [OH] calculation in atmospheric models as a diagnostic tool. However, uncertainties exist associated with the lack of accuracy in dynamic factors in the simplified analytical expression (Eq. 2), such as vertical and horizontal transport, convective velocity above different vegetation cover, as well as the radiation regime influenced by clouds at high altitudes which are likely to affect the OH concentrations. We also evaluated the predominance of SAMBBA data between 11:00 and 14:00 LT, especially for BG and FP groups, which may alter the distribution of the [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio along the diurnal cycle, and consequently modify the OH estimated. The change of the molar yield of the primary first-generation products of the OH-isoprene oxidation, as a function of NO mixing ratio, is another possible source of uncertainty in the estimation of OH concentrations.
Measurements of CO, NOx, and O3 performed in areas not directly affected by local fire emissions reveal the signature of biomass burning in the chemical composition of the background of the Amazonian atmosphere, due to long-range transport of biomass burning tracers both from Africa and the eastern part of Amazonia. In our analysis, we highlight the importance of photochemical age in areas influenced by biomass burning emission, with distinct results for FP and AP. Fresh smoke plumes had the highest mixing ratios of CO and NOx, highlighting the strength of vertical transport through the detection of biomass burning products in the upper levels (> 1200 m).
Regarding isoprene, the measurements in the BG environment agree with values reported by several studies in pristine areas of the Amazon forest. We found much higher levels of isoprene both in fresh (6 ppbv, 1700–2000 m) and aged (4 ppbv, 1600–2000 m) smoke plumes. These results reinforce the hypothesis that fire activity has energy enough to promote the isoprene transport to higher altitudes, altering the isoprene oxidation mechanism when compared with remote areas. Fresh plumes also presented a higher [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio (7.0), when compared with both AP (2.3) and BG (1.7), indicating a strong oxidation process within the boundary layer. Using the complementary approach of the simplified sequential reaction model used by Karl et al. (2007), we indirectly calculated the OH concentration modifying the processing time to represent not only the vertical transport but also the horizontal atmospheric transport time. This adjustment of the processing time provided reasonable OH concentration results close to those obtained in the recent GoAmazon campaign (1 × 106 molecules cm−3).
The highest value for OH in FP within the PBL (1.4 × 106 molecules cm−3) corroborates the results from the [MVK + MACR + ISOPOOH] ∕ [isoprene] ratio, confirming that the photochemical environment in young biomass burning plumes differs from the average conditions. We also detected a strong signal in the oxidative capacity at higher levels (∼ 1000 m), characteristic of the CL existence, as reported by other studies (Karl et al., 2007; Mauldin et al., 1997).
For future research, we recommend further investigation of the impact of the dynamic factors in the estimation of OH mixing ratios, such as horizontal transport and convective velocity above different vegetation cover, as well as the effect of the radiation regime influenced by clouds at high altitudes altering photolysis rates. Considering the recent updates in the molar yield change of the primary first-generation products of the OH-isoprene oxidation, we also expect a reduction in the uncertainties associated with the estimation of OH mixing ratio.
The SAMBBA field experiment data are available at the Centre for Environmental Data Analysis (http://browse.ceda.ac.uk/browse/badc/sambba/data/faam-bae146, Centre for Environmental Data Analysis, 2018) and complementary data are available on request.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-12715-2018-supplement.
FCdS, KML, and ABG prepared the manuscript. FCdS analyzed the chemistry data and performed the estimation of OH density with contributions from KML, ABG, and SK. DG provided the WRF-Chem data used to calculate the estimated OH density. JRH provided isoprene data from the whole air sampling (WAS) system canisters. JL provided NOx data. DEO and GF provided data from isoprene, methyl vinyl ketone, methacrolein, and hydroxy hydroperoxide. SK, DG, DEO, GF, JL, JRH, JB, and SRF reviewed the manuscript.
The authors declare that they have no conflict of interest.
This article is part of the special issue “South AMerican Biomass Burning Analysis (SAMBBA)”. It is not associated with a conference.
The Facility for Airborne Atmospheric Measurement (FAAM) BAe-146 atmospheric
research aircraft is jointly funded by the Met Office and Natural Environment
Research Council and operated by Directflight Ltd. We would like to thank the
dedicated efforts of FAAM, Directflight, INPE, the University of São
Paulo, and the Brazilian Ministry of Science and Technology in making the
SAMBBA measurement campaign possible. We thank Ben Johnson (Met Office) for
his role in coordinating the SAMBBA campaign. Isoprene data from WAS sample
analysis were provided by James R. Hopkins (National Centre for Atmospheric
Science and University of York). The São Paulo Research Foundation
(FAPESP) supported this work through the projects 2012/13575-9, DR
2012/11676-2 and BEPE 2013/03391-0.
Edited by:
Meinrat O. Andreae
Reviewed by: three anonymous referees
Allan, J. D., Morgan, W. T., Darbyshire, E., Flynn, M. J., Williams, P. I., Oram, D. E., Artaxo, P., Brito, J., Lee, J. D., and Coe, H.: Airborne observations of IEPOX-derived isoprene SOA in the Amazon during SAMBBA, Atmos. Chem. Phys., 14, 11393–11407, https://doi.org/10.5194/acp-14-11393-2014, 2014.
Andreae, M. O., Garstang, M., Gregory, G. L., Harriss, R. C., Pereira, M. C., Sachse, G. W., Setzer, A. W., Talbot, R. W., Torres, A. L., and Wofsy, S. C.: Biomass-Burning Emissions and Associated Haze Layers Over Amazonia, J. Geophys. Res., 93, 1509–1527, 1988.
Andreae, M. O., Anderson, B. E., Blake, D. R., Bradshaw, J. D., Collins, J. E., Gregory, G. L., Sachse, G. W., and Shipham, M. C.: Influence of Plumes from Biomass Burning on Atmospheric Chemistry over the Equatorial and Tropical South-Atlantic during Cite-3, J. Geophys. Res., 99, 12793–12808, https://doi.org/10.1029/94jd00263, 1994.
Andreae, M. O., Artaxo, P., Beck, V., Bela, M., Freitas, S., Gerbig, C., Longo, K., Munger, J. W., Wiedemann, K. T., and Wofsy, S. C.: Carbon monoxide and related trace gases and aerosols over the Amazon Basin during the wet and dry seasons, Atmos. Chem. Phys., 12, 6041–6065, https://doi.org/10.5194/acp-12-6041-2012, 2012.
Apel, E. C.: Measurement and interpretation of isoprene fluxes and isoprene, methacrolein, and methyl vinyl ketone mixing ratios at the PROPHET site during the 1998 Intensive, J. Geophys. Res., 107, 1–15, https://doi.org/10.1029/2000JD000225, 2002.
Artaxo, P., Rizzo, L. V, Brito, J. F., Barbosa, H. M. J., Arana, A., Sena, E. T., Cirino, G. G., Bastos, W., Martin, S. T., and Andreae, M. O.: Atmospheric aerosols in Amazonia and land use change: from natural biogenic to biomass burning conditions, Faraday Discuss., 165, 203–235, https://doi.org/10.1039/C3FD00052D, 2013.
Barket, D. J., Grossenbacher, J. W., Hurst, J. M., Shepson, P. B., Olszyna, K., Thornberry, T., Carroll, M. A., Roberts, J., Stroud, C., Bottenheim, J., and Biesenthal, T.: A study of the NOx dependence of isoprene oxidation, J. Geophys. Res.-Atmos., 109, 1–12, https://doi.org/10.1029/2003JD003965, 2004.
Bela, M. M., Longo, K. M., Freitas, S. R., Moreira, D. S., Beck, V., Wofsy, S. C., Gerbig, C., Wiedemann, K., Andreae, M. O., and Artaxo, P.: Ozone production and transport over the Amazon Basin during the dry-to-wet and wet-to-dry transition seasons, Atmos. Chem. Phys., 15, 757–782, https://doi.org/10.5194/acp-15-757-2015, 2015.
Bernhammer, A.-K., Breitenlechner, M., Keutsch, F. N., and Hansel, A.: Technical note: Conversion of isoprene hydroxy hydroperoxides (ISOPOOHs) on metal environmental simulation chamber walls, Atmos. Chem. Phys., 17, 4053–4062, https://doi.org/10.5194/acp-17-4053-2017, 2017.
Bertschi, I., Yokelson, R. J., Ward, D. E., Babbitt, R. E., Susott, R. A., Goode, J. G., and Hao, W. M.: Trace gas and particle emissions from fires in large diameter and belowground biomass fuels, J. Geophys. Res., 108, 8472, https://doi.org/10.1029/2002JD002100, 2003.
Blake, N. J., Blake, D. J., Sive, B. C., Chen, T.-Y., Rowland, F. S., Collins, J. E., Sachse, G. W., and Anderson, B. E.: Biomass burning emissions and vertical distribution of atmospheric methyl halides and other reduced carbon gases in the South Atlantic region, J. Geophys. Res., 101, 151–164, 1996.
Centre for Environmental Data Analysis: South AMerican Biomass Burning Analysis (SAMBBA), available at: http://data.ceda.ac.uk/badc/sambba/, last access: 3 August 2018.
Ciccioli, P., Centritto, M., and Loreto, F.: Biogenic volatile organic compound emissions from vegetation fires, Plant Cell Env., 37, 1810–1825, https://doi.org/10.1111/pce.12336, 2014.
Crutzen, P. J., Delany, A. C., Greenberg, J., Haagenson, P., Heidt, L., Lueb, R., Pollock, W., Seiler, W., Wartburg, A., and Zimmerman, P.: Tropospheric Chemical-Composition Measurements in Brazil During the Dry Season, J. Atmos. Chem., 2, 233–256, 1985.
Dreyfus, G. B., Schade, G. W., and Goldstein, A. H.: Observational constraints on the contribution of isoprene oxidation to ozone production on the western slope of the Sierra Nevada, California, J. Geophys. Res.-Atmos., 107, 1–17, https://doi.org/10.1029/2001JD001490, 2002.
Elshorbany, Y. F., Kurtenbach, R., Wiesen, P., Lissi, E., Rubio, M., Villena, G., Gramsch, E., Rickard, A. R., Pilling, M. J., and Kleffmann, J.: Oxidation capacity of the city air of Santiago, Chile, Atmos. Chem. Phys., 9, 2257–2273, https://doi.org/10.5194/acp-9-2257-2009, 2009.
Feiner, P. A., Brune, W. H., Miller, D. O., Zhang, L., Cohen, R. C., Romer, P. S., Goldstein, A. H., Keutsch, F. N., Skog, K. M., Wennberg, P. O., Nguyen, T. B., Teng, A. P., DeGouw, J., Koss, A., Wild, R. J., Brown, S. S., Guenther, A., Edgerton, E., Baumann, K., and Fry, J. L.: Testing Atmospheric Oxidation in an Alabama Forest, J. Atmos. Sci., 73, 4699–4710, https://doi.org/10.1175/JAS-D-16-0044.1, 2016.
Freitas, S. R., Longo, K. M., and Andreae, M. O.: Impact of including the plume rise of vegetation fires in numerical simulations of associated atmospheric pollutants, Geophys. Res. Lett., 33, 951–955, https://doi.org/10.1029/2006GL026608, 2006.
Freitas, S. R., Longo, K. M., Chatfield, R., Latham, D., Silva Dias, M. A. F., Andreae, M. O., Prins, E., Santos, J. C., Gielow, R., and Carvalho Jr., J. A.: Including the sub-grid scale plume rise of vegetation fires in low resolution atmospheric transport models, Atmos. Chem. Phys., 7, 3385–3398, https://doi.org/10.5194/acp-7-3385-2007, 2007.
Freitas, S. R., Longo, K. M., Trentmann, J., and Latham, D.: Technical Note: Sensitivity of 1-D smoke plume rise models to the inclusion of environmental wind drag, Atmos. Chem. Phys., 10, 585–594, https://doi.org/10.5194/acp-10-585-2010, 2010.
Gerbig, C., Kley, D., Volz-thomas, A., Kent, J., Dewey, K., and Mckenna, D. S.: Fast response resonance fluorescence CO measurements aboard the C-130: Instrument characterization and measurements made during North Atlantic Regional Experiment 1993, J. Geophys. Res., 101, 29229–29238, 1996.
Gerbig, C., Schmitgen, S., Kley, D., Volz-thomas, A., and Dewey, K.: An improved fast-response vacuum-UV resonance fluorescence CO instrument, J. Geophys. Res., 104, 1699–1704, 1999.
Greenberg, J. P. and Zimmerman, P. R.: Nonmethane hydrocarbons in remote tropical, continental, and marine atmospheres, J. Geophys. Res., 89, 4767, https://doi.org/10.1029/JD089iD03p04767, 1984.
Greenberg, J. P., Zimmerman, P. R., Greenberg, J. P., and Westberg, C. E.: Measurements of Atmospheric Hydrocarbons and Biogenic Emission Fluxes in the Amazon Boundary Layer Trade Wind Convective, J. Geophys. Res.-Atmos., 93, 1407–1416, https://doi.org/10.1029/JD093iD02p01407, 1988.
Greenberg, J. P., Guenther, A. B., Petron, G., Wiedinmyer, C., Vega, O., Gatti, L. V., Tota, J., and Fisch, G.: Biogenic VOC emissions from forested Amazonian landscapes, Glob. Chang. Biol., 10, 651–662, https://doi.org/10.1111/j.1365-2486.2004.00758.x, 2004.
Gregory, G. L., Harriss, R. C., Talbot, R. W., Rasmussen, R. A., Garstang, M., Andreae, M. O., Hinton, R. R., Browell, E. V., Beck, S. M., Sebacher, D. I., Khalil, M. A. K., Ferek, R. J., and Harriss, S. V.: Air chemistry over the tropical forest of Guyana, J. Geophys. Res., 91, 8603–8612, 1986.
Guenther, A., Karl, T., Harley, P., Wiedinmyer, C., Palmer, P. I., and Geron, C.: Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature), Atmos. Chem. Phys., 6, 3181–3210, https://doi.org/10.5194/acp-6-3181-2006, 2006.
Harley, P., Vasconcellos, P., Vierling, L., Pinheiro, C. C. D. S. ., Greenberg, J., Guenther, A., Klinger, L., de Almeida, S. S., Neill, D., Baker, T., Phillips, O., Malhi, Y., Division, A. C., City, R., Louis, S., and Management, R.: Variation in potential for isoprene emissions among Neotropical forest sites, Glob. Chang. Biol., 10, 630–650, https://doi.org/10.1111/j.1529-8817.2003.00760.x, 2004.
Helmig, D., Balsley, B., Davis, K., Kuck, L. R., Jensen, M., Bognar, J., Smith, T., Arrieta, R. V., Rodríguez, R., and Birks, J. W.: Vertical profiling and determination of landscape fluxes of biogenic nonmethane hydrocarbons within the planetary boundary layer in the Peruvian Amazon, J. Geophys. Res.-Atmos., 103, 25519–25532, https://doi.org/10.1029/98jd01023, 1998.
Hobbs, P. V., Sinha, P., Yokelson, R. J., Christian, T. J., Blake, D. R., Gao, S., Kirchstetter, T. W., Novakov, T., and Pilewskie, P.: Evolution of gases and particles from a savanna fire in South Africa, J. Geophys. Res., 108, 8485, https://doi.org/10.1029/2002JD002352, 2003.
Hopkins, J. R., Jones, C. E., and Lewis, A. C.: A dual channel gas chromatograph for atmospheric analysis of volatile organic compounds including oxygenated and monoterpene compounds, J. Environ. Monit., 13, 2268–2276, https://doi.org/10.1039/c1em10050e, 2011.
Jacob, D. J. and Wofsy, S. C.: Photochemistry of biogenic emissions over the Amazon forest, J. Geophys. Res.-Atmos., 93, 1477–1486, https://doi.org/10.1029/JD093iD02p01477, 1988.
Janssens-Maenhout, G., Crippa, M., Guizzardi, D., Dentener, F., Muntean, M., Pouliot, G., Keating, T., Zhang, Q., Kurokawa, J., Wankmüller, R., Denier van der Gon, H., Kuenen, J. J. P., Klimont, Z., Frost, G., Darras, S., Koffi, B., and Li, M.: HTAP_v2.2: a mosaic of regional and global emission grid maps for 2008 and 2010 to study hemispheric transport of air pollution, Atmos. Chem. Phys., 15, 11411–11432, https://doi.org/10.5194/acp-15-11411-2015, 2015.
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015.
Jost, C., Trentmann, J., Sprung, D., and Andreae, M.: Trace gas chemistry in a young biomass burning plume over Namibia: Observations and model simulations, J. Geophys. Res., 108, 8482, https://doi.org/10.1029/2002JD002431, 2003.
Karl, T., Guenther, A., Yokelson, R. J., Greenberg, J., Potosnak, M., Blake, D. R., and Artaxo, P.: The tropical forest and fire emissions experiment: Emission, chemistry, and transport of biogenic volatile organic compounds in the lower atmosphere over Amazonia, J. Geophys. Res., 112, D18302, https://doi.org/10.1029/2007JD008539, 2007.
Kaufman, Y. J., Setzer, A., Ward, D., Tanre, D., Holben, B. N., Menzel, P., Pereira, M. C., and Rasmussen, R.: Biomass Burning Airborne and Spaceborne Experiment in the Amazonas (Base-a), J. Geophys. Res., 97, 14581–14599, https://doi.org/10.1029/92JD00275, 1992.
Kubistin, D., Harder, H., Martinez, M., Rudolf, M., Sander, R., Bozem, H., Eerdekens, G., Fischer, H., Gurk, C., Klüpfel, T., Königstedt, R., Parchatka, U., Schiller, C. L., Stickler, A., Taraborrelli, D., Williams, J., and Lelieveld, J.: Hydroxyl radicals in the tropical troposphere over the Suriname rainforest: comparison of measurements with the box model MECCA, Atmos. Chem. Phys., 10, 9705–9728, https://doi.org/10.5194/acp-10-9705-2010, 2010.
Kuhn, U., Andreae, M. O., Ammann, C., Araújo, A. C., Brancaleoni, E., Ciccioli, P., Dindorf, T., Frattoni, M., Gatti, L. V., Ganzeveld, L., Kruijt, B., Lelieveld, J., Lloyd, J., Meixner, F. X., Nobre, A. D., Pöschl, U., Spirig, C., Stefani, P., Thielmann, A., Valentini, R., and Kesselmeier, J.: Isoprene and monoterpene fluxes from Central Amazonian rainforest inferred from tower-based and airborne measurements, and implications on the atmospheric chemistry and the local carbon budget, Atmos. Chem. Phys., 7, 2855–2879, https://doi.org/10.5194/acp-7-2855-2007, 2007.
Langford, A. O., Senff, C. J., Alvarez II, R. J., Banta, R. M., Hardesty, R. M., Kleffmann, J., Gavriloaiei, T., Hofzumahaus, A., Holland, F., Koppmann, R., Rupp, L., Schlosser, E., Siese, M., and Wahner, A.: Daytime formation of nitrous acid: A major source of OH radicals in a forest, Geophys. Res. Lett., 32, 1–4, https://doi.org/10.1029/2005GL022524, 2005.
Laothawornkitkul, J., Taylor, J. E., Paul, N. D., and Hewitt, C. N.: Biogenic volatile organic compounds in the Earth system, New Phytol., 183, 27–51, https://doi.org/10.1111/j.1469-8137.2009.02859.x, 2009.
Lelieveld, J., Butler, T. M., Crowley, J. N., Dillon, T. J., Fischer, H., Ganzeveld, L., Harder, H., Lawrence, M. G., Martinez, M., Taraborrelli, D., and Williams, J.: Atmospheric oxidation capacity sustained by a tropical forest, Nature, 452, 737–740, https://doi.org/10.1038/nature06870, 2008.
Lew, M. M., Dusanter, S., and Stevens, P. S.: Measurement of interferences associated with the detection of the hydroperoxy radical in the atmosphere using laser-induced fluorescence, Atmos. Meas. Tech., 11, 95–109, https://doi.org/10.5194/amt-11-95-2018, 2018.
Liu, Y. J., Herdlinger-Blatt, I., McKinney, K. A., and Martin, S. T.: Production of methyl vinyl ketone and methacrolein via the hydroperoxyl pathway of isoprene oxidation, Atmos. Chem. Phys., 13, 5715–5730, https://doi.org/10.5194/acp-13-5715-2013, 2013.
Liu, Y., Brito, J., Dorris, M., Rivera-Rios, J. C., Seco, R., Bates, K. H., Artaxo, P., Junior, S. D., Keutsch, F., Kim, S., Goldstein, A. H., Guenther, A. B., Manzi, A., Souza, R., Springston, S. R., Watson, T. B., McKinney, K. A., and Martin, S. T.: Isoprene Photochemistry over the Amazon Rain Forest, P. Natl. Acad. Sci. USA, 113, 6125–6130, https://doi.org/10.1073/pnas.1524136113, 2016.
Logan, J. A., Prather, M. J., Wofsy, S. C., and McElroy, M. B.: Tropospheric chemistry: A global perspective, J. Geophys. Res., 86, 7210–7254, https://doi.org/10.1029/JC086iC08p07210, 1981.
Longo, K. M., Freitas, S. R., Andreae, M. O., Yokelson, R., and Artaxo, P.: Biomass Burning in Amazonia: Emissions, Long-Range Transport of Smoke and Its Regional and Remote Impacts, in: Amazonia and Global Change, edited by: Keller, M., Bustamante, M., Gash, J., and Silva Dias, P., https://doi.org/10.1029/2008GM000717, 2013.
Lu, K. D., Rohrer, F., Holland, F., Fuchs, H., Bohn, B., Brauers, T., Chang, C. C., Häseler, R., Hu, M., Kita, K., Kondo, Y., Li, X., Lou, S. R., Nehr, S., Shao, M., Zeng, L. M., Wahner, A., Zhang, Y. H., and Hofzumahaus, A.: Observation and modelling of OH and HO2 concentrations in the Pearl River Delta 2006: a missing OH source in a VOC rich atmosphere, Atmos. Chem. Phys., 12, 1541–1569, https://doi.org/10.5194/acp-12-1541-2012, 2012.
Mao, J., Ren, X., Zhang, L., Van Duin, D. M., Cohen, R. C., Park, J.-H., Goldstein, A. H., Paulot, F., Beaver, M. R., Crounse, J. D., Wennberg, P. O., DiGangi, J. P., Henry, S. B., Keutsch, F. N., Park, C., Schade, G. W., Wolfe, G. M., Thornton, J. A., and Brune, W. H.: Insights into hydroxyl measurements and atmospheric oxidation in a California forest, Atmos. Chem. Phys., 12, 8009–8020, https://doi.org/10.5194/acp-12-8009-2012, 2012.
Mauldin, R. L. I., Madronich, S., Flocke, S. J., Eisele, F. L., Frost, G. J., and Prevot, A. S. H.: New insights on OH: Measurements around and in clouds, Geophys. Res. Lett., 24, 3033–3036, https://doi.org/10.1029/97GL02983, 1997.
Mauzerall, D. L., Logan, J. A., Jacob, D. J., Anderson, B. E., Blake, D. R., Bradshaw, J. D., Heikes, B., Sachse, G. W., Singh, H., and Talbot, B.: Photochemistry in biomass burning plumes and implications for tropospheric ozone over the tropical South Atlantic, J. Geophys. Res., 103, 8401, https://doi.org/10.1029/97JD02612, 1998.
Monks, P. S.: Gas-phase radical chemistry in the troposphere, Chem. Soc. Rev., 34, 376–395, https://doi.org/10.1039/b307982c, 2005.
Müller, M., Anderson, B. E., Beyersdorf, A. J., Crawford, J. H., Diskin, G. S., Eichler, P., Fried, A., Keutsch, F. N., Mikoviny, T., Thornhill, K. L., Walega, J. G., Weinheimer, A. J., Yang, M., Yokelson, R. J., and Wisthaler, A.: In situ measurements and modeling of reactive trace gases in a small biomass burning plume, Atmos. Chem. Phys., 16, 3813–3824, https://doi.org/10.5194/acp-16-3813-2016, 2016.
Murphy, J. G., Oram, D. E., and Reeves, C. E.: Measurements of volatile organic compounds over West Africa, Atmos. Chem. Phys., 10, 5281–5294, https://doi.org/10.5194/acp-10-5281-2010, 2010.
Nguyen, T. B., Crounse, J. D., Schwantes, R. H., Teng, A. P., Bates, K. H., Zhang, X., St. Clair, J. M., Brune, W. H., Tyndall, G. S., Keutsch, F. N., Seinfeld, J. H., and Wennberg, P. O.: Overview of the Focused Isoprene eXperiment at the California Institute of Technology (FIXCIT): mechanistic chamber studies on the oxidation of biogenic compounds, Atmos. Chem. Phys., 14, 13531–13549, https://doi.org/10.5194/acp-14-13531-2014, 2014.
Palmer, P. I., Parrington, M., Lee, J. D., Lewis, A. C., Rickard, A. R., Bernath, P. F., Duck, T. J., Waugh, D. L., Tarasick, D. W., Andrews, S., Aruffo, E., Bailey, L. J., Barrett, E., Bauguitte, S. J.-B., Curry, K. R., Di Carlo, P., Chisholm, L., Dan, L., Forster, G., Franklin, J. E., Gibson, M. D., Griffin, D., Helmig, D., Hopkins, J. R., Hopper, J. T., Jenkin, M. E., Kindred, D., Kliever, J., Le Breton, M., Matthiesen, S., Maurice, M., Moller, S., Moore, D. P., Oram, D. E., O'Shea, S. J., Owen, R. C., Pagniello, C. M. L. S., Pawson, S., Percival, C. J., Pierce, J. R., Punjabi, S., Purvis, R. M., Remedios, J. J., Rotermund, K. M., Sakamoto, K. M., da Silva, A. M., Strawbridge, K. B., Strong, K., Taylor, J., Trigwell, R., Tereszchuk, K. A., Walker, K. A., Weaver, D., Whaley, C., and Young, J. C.: Quantifying the impact of BOReal forest fires on Tropospheric oxidants over the Atlantic using Aircraft and Satellites (BORTAS) experiment: design, execution and science overview, Atmos. Chem. Phys., 13, 6239–6261, https://doi.org/10.5194/acp-13-6239-2013, 2013.
Parrish, D. D., Holloway, J. S., Trainer, M., Murphy, P. C., Fehsenfeld, F. C., and Forbes, G. L.: Export of North American Ozone Pollution to the North Atlantic Ocean, Science, 259, 1436–1439, https://doi.org/10.1126/science.259.5100.1436, 1993.
Prinn, R. G.: Treatise on Geochemistry: Ozone, Hydroxyl Radical, and Oxidative Capacity, 2nd ed., Massachusetts Institute of Technology, Cambridge, MA, USA, 1–18, 2014.
Rasmussen, R. A. and Khalil, M. A. K.: Isoprene over the Amazon Basin, J. Geophys. Res., 93, 1417–1421, https://doi.org/10.1029/JD093iD02p01417, 1988.
Reid, J. S., Hobbs, P. V., Ferek, R. J., Blake, D. R., Martins, J. V., Dunlap, M. R., and Liousse, C.: Physical, chemical, and optical properties of regional hazes dominated by smoke in Brazil, J. Geophys. Res., 103, 32059–32080, 1998.
Rivera-Rios, J. C., Nguyen, T. B., Crounse, J. D., Jud, W., St. Clair, J. M., Mikoviny, T., Gilman, J. B., Lerner, B. M., Kaiser, J. B., De Gouw, J., Wisthaler, A., Hansel, A., Wennberg, P. O., Seinfeld, J. H., Keutsch, F. N., Lin, G., Penner, J. E., and Zhou, C.: Conversion of hydroperoxides to carbonyls in field and laboratory instrumentation: Observational bias in diagnosing pristine versus anthropogenically controlled atmospheric chemistry, Geophys. Res. Lett., 41, 8645–8651, https://doi.org/10.1002/2014GL061919, 2014.
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kurten, A., St Clair, J. M., Seinfeld, J. H., and Wennberg, P. O.: Unexpected epoxide formation in the gas-phase photooxidation of isoprene, Science, 325, 730–733, 2009.
Rohrer, F., Lu, K., Hofzumahaus, A., Bohn, B., Brauers, T., Chang, C.-C., Fuchs, H., Haeseler, R., Holland, F., Hu, M., Kita, K., Kondo, Y., Li, X., Lou, S., Oebel, A., Shao, M., Zeng, L., Zhu, T., Zhang, Y., and Wahner, A.: Maximum efficiency in the hydroxyl-radical-based self-cleansing of the troposphere, Nat. Geosci., 7, 559–563, https://doi.org/10.1038/NGEO2199, 2014.
Sharkey, T. D., Wiberley, A. E., and Donohue, A. R.: Isoprene emission from plants: Why and how, Ann. Bot., 101, 5–18, 2008.
Singh, H. B., Herlth, D., O'Hara, D., Salas, L., Torres, A. L., Gregory, G. L., Sachse, G. W., and Kasting, J. F.: Atmospheric peroxyacetyl nitrate measurements over the Brazilian Amazon Basin during the wet season: Relationships with nitrogen oxides and ozone, J. Geophys. Res., 95, 945–954, 1990.
Ter Steege, H., Pitman, N. C. A., Sabatier, D., Baraloto, C., Salomão, R. P., Guevara, J. E., Phillips, O. L., Castilho, C. V., Magnusson, W. E., Molino, J. F., Monteagudo, A., Núñez Vargas, P., Montero, J. C., Feldpausch, T. R., Coronado, E. N. H., Killeen, T. J., Mostacedo, B., Vasquez, R., Assis, R. L., Terborgh, J., Wittmann, F., Andrade, A., Laurance, W. F., Laurance, S. G. W., Marimon, B. S., Marimon, B. H., Guimarães Vieira, I. C., Amaral, I. L., Brienen, R., Castellanos, H., Cárdenas López, D., Duivenvoorden, J. F., Mogollón, H. F., Matos, F. D. A., Dávila, N., García-Villacorta, R., Stevenson Diaz, P. R., Costa, F., Emilio, T., Levis, C., Schietti, J., Souza, P., Alonso, A., Dallmeier, F., Montoya, A. J. D., Fernandez Piedade, M. T., Araujo-Murakami, A., Arroyo, L., Gribel, R., Fine, P. V. A., Peres, C. A., Toledo, M., Aymard C, G. A., Baker, T. R., Cerón, C., Engel, J., Henkel, T. W., Maas, P., Petronelli, P., Stropp, J., Zartman, C. E., Daly, D., Neill, D., Silveira, M., Paredes, M. R., Chave, J., Lima Filho, D. A., Jørgensen, P. M., Fuentes, A., Schöngart, J., Cornejo Valverde, F., Di Fiore, A., Jimenez, E. M., Peñuela Mora, M. C., Phillips, J. F., Rivas, G., van Andel, T. R., von Hildebrand, P., Hoffman, B., Zent, E. L., Malhi, Y., Prieto, A., Rudas, A., Ruschel, A. R., Silva, N., Vos, V. A., Zent, S., Oliveira, A. A., Schutz, A. C., Gonzales, T., Trindade Nascimento, M., Ramirez-Angulo, H., Sierra, R., Tirado, M., Umaña Medina, M. N., van der Heijden, G., Vela, C. I. A., Vilanova Torre, E., Vriesendorp, C., et al.: Hyperdominance in the Amazonian tree flora, Science, 80, 342, 1243092, https://doi.org/10.1126/science.1243092, 2013.
Stroud, C. A., Roberts, J. M., Goldan, P. D., Kuster, W. C., Murphy, P. C., Williams, E. J., Hereid, D., Parrish, D., Sueper, D., Trainer, M., Fehsenfeld, F. C., Apel, E. C., Riemer, D., Wert, B., Henry, B., Fried, A., Martinez-Harder, M., Harder, H., Brune, W. H., Li, G., Xie, H., and Young, V. L.: Isoprene and its oxidation products, methacrolein and methylvinyl ketone, at an urban forested site during the 1999 Southern Oxidants Study, J. Geophys. Res.-Atmos., 106, 8035–8046, https://doi.org/10.1029/2000JD900628, 2001.
Teng, A. P., Crounse, J. D., and Wennberg, P. O.: Isoprene Peroxy Radical Dynamics, J. Am. Chem. Soc., 139, 5367–5377, https://doi.org/10.1021/jacs.6b12838, 2017.
Torres, A. L. and Buchan, H.: Tropospheric nitric oxide measurements over the Amazon Basin, J. Geophys. Res.-Atmos., 93, 1396–1406, https://doi.org/10.1029/JD093iD02p01396, 1988.
Trentmann, J. and Andreae, M.: Chemical processes in a young biomass-burning plume, J. Geophys. Res., 108, 4705, https://doi.org/10.1029/2003JD003732, 2003.
Warneke, C., Holzinger, R., Hansel, A., Jordan, A., Lindinger, W., Poschl, U., Williams, J., Hoor, P., Fischer, H., Crutzen, P. J., Scheeren, H. A., Lelieveld, J., Pöschl, U., Williams, J., Hoor, P., Fischer, H., Crutzen, P. J., Scheeren, H. A., and Lelieveld, J.: Isoprene and Its Oxidation ProductsMethyl Vinyl Ketone, Methacrolein, and Isoprene Related Peroxides Measured Online over the Tropical Rain Forest of Surinam in March 1998, J. Atmos. Chem., 38, 167–185, https://doi.org/10.1023/A:1006326802432, 2001.
Whalley, L. K., Edwards, P. M., Furneaux, K. L., Goddard, A., Ingham, T., Evans, M. J., Stone, D., Hopkins, J. R., Jones, C. E., Karunaharan, A., Lee, J. D., Lewis, A. C., Monks, P. S., Moller, S. J., and Heard, D. E.: Quantifying the magnitude of a missing hydroxyl radical source in a tropical rainforest, Atmos. Chem. Phys., 11, 7223–7233, https://doi.org/10.5194/acp-11-7223-2011, 2011.
Whalley, L., Stone, D., and Heard D.: New Insights into the Tropospheric Oxidation of Isoprene: Combining Field Measurements, Laboratory Studies, Chemical Modelling and Quantum Theory, in: Atmospheric and Aerosol Chemistry, Topics in Current Chemistry, edited by: McNeill, V. and Ariya, P., vol. 339, Springer, Berlin, Heidelberg, https://doi.org/10.1007/128_2012_359, 2012.
Williams, J., Poschl, U., Crutzen, P. J., Hansel, A., Holzinger, R., Warneke, C., Lindinger, W., and Lelieveld, J.: An atmospheric chemistry interpretation of mass scans obtained from a proton transfer mass spectrometer flown over the tropical rainforest of Surinam, J. Atmos. Chem., 38, 133–166, https://doi.org/10.1023/A:1006322701523, 2001.
Yáñez-Serrano, A. M., Nölscher, A. C., Williams, J., Wolff, S., Alves, E., Martins, G. A., Bourtsoukidis, E., Brito, J., Jardine, K., Artaxo, P., and Kesselmeier, J.: Diel and seasonal changes of biogenic volatile organic compounds within and above an Amazonian rainforest, Atmos. Chem. Phys., 15, 3359–3378, https://doi.org/10.5194/acp-15-3359-2015, 2015.
Yokelson, R., Bertschi, I., Christian, T., Hobbs, P., Ward, D., and Hao, W.: Trace gas measurements in nascent, aged, and cloud-processed smoke from African savanna fires by airborne Fourier transform infrared spectroscopy (AFTIR), J. Geophys. Res.-Atmos., 108, 1–18, https://doi.org/10.1029/2002JD002322, 2003.
Yokelson, R. J., Karl, T., Artaxo, P., Blake, D. R., Christian, T. J., Griffith, D. W. T., Guenther, A., and Hao, W. M.: The Tropical Forest and Fire Emissions Experiment: overview and airborne fire emission factor measurements, Atmos. Chem. Phys., 7, 5175–5196, https://doi.org/10.5194/acp-7-5175-2007, 2007.
Zimmerman, P. R., Greenberg, J. P., and Westberg, C. E.: Measurements of atmospheric hydrocarbons and biogenic emission fluxes in the Amazon Boundary layer, J. Geophys. Res., 93, 1407, https://doi.org/10.1029/jd093id02p01407, 1988.