the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 30 Jul 2018
| 30 Jul 2018
Aircraft observations of the chemical composition and aging of aerosol in the Manaus urban plume during GoAmazon 2014/5
Mikhail S. Pekour
Edward C. Fortner
Paulo Artaxo
Suzane de Sá
John M. Hubbe
Karla M. Longo
Luiz A. T. Machado
Scot T. Martin
Stephen R. Springston
Jason Tomlinson
Jian Wang
The Green Ocean Amazon (GoAmazon 2014/5) campaign, conducted from January 2014 to December 2015 in the vicinity of Manaus, Brazil, was designed to study the aerosol life cycle and aerosol–cloud interactions in both pristine and anthropogenically influenced conditions. As part of this campaign, the U.S. Department of Energy (DOE) Gulfstream 1 (G-1) research aircraft was deployed from 17 February to 25 March 2014 (wet season) and 6 September to 5 October 2014 (dry season) to investigate aerosol and cloud properties aloft. Here, we present results from the G-1 deployments focusing on measurements of the aerosol chemical composition and secondary organic aerosol (SOA) formation and aging.
In the first portion of the paper, we provide an overview of the data and compare and contrast the data from the wet and dry season. Organic aerosol (OA) dominates the deployment-averaged chemical composition, comprising 80 % of the non-refractory PM1 aerosol mass, with sulfate comprising 14 %, nitrate 2 %, and ammonium 4 %. This product distribution was unchanged between seasons, despite the fact that total aerosol loading was significantly higher in the dry season and that regional and local biomass burning was a significant source of OA mass in the dry, but not wet, season. However, the OA was more oxidized in the dry season, with the median of the mean carbon oxidation state increasing from −0.45 in the wet season to −0.02 in the dry season.
In the second portion of the paper, we discuss the evolution of the Manaus plume, focusing on 13 March 2014, one of the exemplary days in the wet season. On this flight, we observe a clear increase in OA concentrations in the Manaus plume relative to the background. As the plume is transported downwind and ages, we observe dynamic changes in the OA. The mean carbon oxidation state of the OA increases from −0.6 to −0.45 during the 4–5 h of photochemical aging. Hydrocarbon-like organic aerosol (HOA) mass is lost, with ΔHOA∕ΔCO values decreasing from 17.6 µg m−3 ppmv−1 over Manaus to 10.6 µg m−3 ppmv−1 95 km downwind. Loss of HOA is balanced out by formation of oxygenated organic aerosol (OOA), with ΔOOA∕ΔCO increasing from 9.2 to 23.1 µg m−3 ppmv−1. Because hydrocarbon-like organic aerosol (HOA) loss is balanced by OOA formation, we observe little change in the net Δorg∕ΔCO values; Δorg∕ΔCO averages 31 µg m−3 ppmv−1 and does not increase with aging. Analysis of the Manaus plume evolution using data from two additional flights in the wet season showed similar trends in Δorg∕ΔCO to the 13 March flight; Δorg∕ΔCO values averaged 34 µg m−3 ppmv−1 and showed little change over 4–6.5 h of aging. Our observation of constant Δorg∕ΔCO are in contrast to literature studies of the outflow of several North American cities, which report significant increases in Δorg∕ΔCO for the first day of plume aging. These observations suggest that SOA formation in the Manaus plume occurs, at least in part, by a different mechanism than observed in urban outflow plumes in most other literature studies. Constant Δorg∕ΔCO with plume aging has been observed in many biomass burning plumes, but we are unaware of reports of fresh urban emissions aging in this manner. These observations show that urban pollution emitted from Manaus in the wet season forms less particulate downwind as it ages than urban pollution emitted from North American cities.
- Article
(13794 KB) -
Supplement
(827 KB) - BibTeX
- EndNote





Aerosol particles have important impacts on visibility, human health, and the Earth's energy balance and water cycle. The impact of aerosol particles on radiation balance, in particular their impact on cloud properties and lifetimes, continues to be a significant source of uncertainty for global climate models (IPCC, 2013). An extensive series of field studies has shown that a large fraction of the total non-refractory aerosol mass is organic aerosol (OA) and that a large fraction of this OA mass forms in the atmosphere when organic compounds in the gas phase are oxidized and subsequently condense as secondary organic aerosol (SOA) (Zhang et al., 2007; Jimenez et al., 2009). Because SOA is such a large fraction of the aerosol mass, condensation of SOA is critical to growing nucleation-mode particles, which are initially too small to serve as cloud condensation nuclei (CCN), to sizes that are capable of forming cloud droplets (Ehn et al., 2014; Riipinen et al., 2011; Pierce et al., 2012), though a recent study showed that unique conditions in the Amazon allowed particles smaller than 50 nm to act as CCN during Green Ocean Amazon campaign (GoAmazon 2014/5; Fan et al., 2018). Thus, accurate descriptions of SOA condensation and aerosol growth kinetics are crucial to accurately predicting aerosol size distributions and therefore CCN number concentrations and aerosol optical properties, both of which are required for accurately predicting the impact of aerosols on climate (Scott et al., 2015; Riipinen et al., 2012; Zaveri et al., 2014).
For several years, there has been an interest in studying the Lagrangian (i.e., within the same air parcel) evolution of organic aerosol from the emissions of urban centers. Field studies have investigated the evolution of pollution plumes by arranging fixed observation sites at different distances downwind of a city along the direction of the prevailing wind or by tracking the plume with a mobile platform, such as an aircraft. Larger campaigns may employ both strategies. Most studies of this type are best described as pseudo-Lagrangian as repeatedly sampling the same air parcel is difficult with mobile platforms and impossible with fixed sites. Dilution and mixing of the air parcel with background air also alter the plume composition. To account for atmospheric dilution and spatial and temporal variability in emissions, studies often utilize the ratio of excess OA to that of an inert tracer as a metric for evaluating OA formation in a plume (Kleinman et al., 2008; Sullivan and Weber, 2006; Takegawa et al., 2006; de Gouw et al., 2005). CO is a common choice for the inert tracer because it is emitted during combustion and other anthropogenic processes, is significantly enhanced in urban plumes relative to the background, and is routinely and robustly measured. Measurements of Δorg∕ΔCO in aged urban outflow have spanned a range from 47 µg m−3 ppmv−1 in the NE USA (de Gouw et al., 2008) to 62–80 µg m−3 ppmv−1 in Mexico City (DeCarlo et al., 2008; Kleinman et al., 2008) to 100 µg m−3 ppmv−1 in the Po Valley, Italy (Crosier et al., 2007), to 44–197 µg m−3 ppmv−1 in Sacramento, CA (Shilling et al., 2013; Setyan et al., 2012), to 97–133 µg m−3 ppmv−1 in Paris (Freney et al., 2014). The same studies have found that Δorg∕ΔCO increases roughly linearly with air mass age for the first 1 day of aging and levels off after approximately 2 days (de Gouw and Jimenez, 2009; DeCarlo et al., 2010; Kleinman et al., 2008; Takegawa et al., 2006; Sullivan et al., 2006; Freney et al., 2014). Observations suggest that changes in Δorg∕ΔCO begin soon after emission. For example, measurements during the MILAGRO campaign showed that Δorg∕ΔCO increased from 10–35 µg m−3 ppmv−1 for fresh emissions to 70–80 µg m−3 ppmv−1 after approximately 1 day of photochemical aging (DeCarlo et al., 2010; Kleinman et al., 2008). Changes of similar magnitude over similar aging times have been measured in the urban outflow of Tokyo (Takegawa et al., 2006), the SE USA (Sullivan et al., 2006), Paris (Freney et al., 2014), and the NE USA (de Gouw et al., 2005).
Measurements have also suggested that organic aerosol production may be enhanced when urban emissions interact with biogenic emissions. Organic aerosol concentrations have shown strong correlation with CO and other tracers of anthropogenic emissions (de Gouw et al., 2005; Volkamer et al., 2006; Weber et al., 2007; Sullivan et al., 2006). Furthermore, satellite observations suggest that the spatial and seasonal patterns of aerosol optical depth coincide with biogenic emissions (Goldstein et al., 2009). At the same time, radiocarbon dating has shown that a large fraction of the carbon in the aerosol phase is modern and that the modern carbon fraction increases with increasing distance from urban centers (Weber et al., 2007; Schichtel et al., 2008). During the CARES campaign near Sacramento, CA, aircraft observations showed that Δorg∕ΔCO measurements were 35–44 µg m−3 ppmv−1 when anthropogenic emissions evolved in the absence of strong biogenic emissions and 77–157 µg m−3 ppmv−1 when the urban plume interacted with regions of strong biogenic emissions (Shilling et al., 2013). Measurements from a ground site during CARES report average Δorg∕ΔCO values of 36 µg m−3 ppmv−1 in the Sacramento plume during periods of low biogenic emissions and 97 µg m−3 ppmv−1 during periods of high biogenic emissions (Setyan et al., 2012). Several additional field studies report enhancements of SOA through interactions of anthropogenic and biogenic emissions but may not employ the Δorg∕ΔCO metric (Zhou et al., 2016; Xu et al., 2015; Ng et al., 2017; Bean et al., 2016). The potential mechanisms responsible for these enhancements are known in some cases but remain uncertain in others and have been outlined in recent review articles (Shrivastava et al., 2017; Ng et al., 2017; Glasius and Goldstein, 2016; Hoyle et al., 2011).
To date, most studies of the evolution of urban plumes have been conducted in the Northern Hemisphere. Far fewer studies have been performed in the Southern Hemisphere, where there is less landmass, population is lower, and therefore background concentrations of anthropogenic pollutants are lower (Andreae, 2007). The Amazon tropical forest, in particular, is an important ecosystem in which background levels of anthropogenic pollution may sometimes reach levels characteristic of pre-industrial conditions but is becoming increasingly impacted by industrialization and anthropogenic pollution (Martin et al., 2017, 2010b; Andreae et al., 2015). Fuzzi et al. (2007) described chemical analysis of filter samples collected in Rondônia, Brazil (∼825 km SW of Manaus, Brazil), from September to November 2002. They found that water-soluble organic species were the main contributors to submicron particles (90 %), with the balance consisting of soluble inorganic ions (Fuzzi et al., 2007). Allan et al. (2014) discuss measurements from flights investigating biogenic processes during the South American Biomass Burning Analysis (SAMBBA) aircraft campaign, based out of Rondônia, Brazil. They report that concentrations of SOA derived from isoprene epoxydiols (IEPOX) were highest in the presence of acidic seed, low NOx concentrations, and high relative humidity (RH; Allan et al., 2014). The AMAZE-08 campaign was conducted in February and March of 2008, and Chen et al. (2009, 2015) reported on chemical composition measurements made with an Aerodyne High-Resolution Time-of-Flight Aerosol Mass Spectrometer located at a tower site 60 km NNW of Manaus, Brazil (Martin et al., 2010a; Chen et al., 2009, 2015). They found that organics accounted for more than 80 % of the non-refractory aerosol mass, with the organic mass often dominated by SOA, and that the aerosol was acidic in composition (Chen et al., 2015). It is important to note that Chen et al. (2009, 2015) excluded data impacted by Manaus emissions from their analysis. De Sá et al. (2017) investigated the production of IEPOX SOA downwind of Manus during GoAmazon2014/5 and found decreased concentrations of IEPOX SOA in the plume relative to the background, which they attribute to suppression of IEPOX formation by elevated NO concentrations in the plume. Kuhn et al. (2010) describe aircraft-based aerosol measurements in the Manaus plume as it was transported downwind. They report increases in ΔCCN∕ΔCO as the plume aged, which they attribute to condensation of both organic and inorganic mass, but did not measure the aerosol chemical composition (Kuhn et al., 2010). Additional aircraft campaigns conducted in the Amazon have often focused on biomass burning and the impact of biomass burning aerosol on cloud properties (Morgan et al., 2013; Yokelson et al., 2007; Andreae et al., 2012).
GoAmazon 2014/5 was conducted in the vicinity of Manaus, Brazil, from January 2014 to December 2015. The goal of GoAmazon 2014/5 was to investigate the interaction of Manaus urban emissions with the surrounding pristine Amazon Basin and the subsequent impact of these emission on cloud formation and properties (Martin et al., 2017, 2016). Manaus's geography makes it ideal for this mission. Manaus, an industrial city with a metropolitan population of more than 2 million people, is the largest city in the Amazon Basin. The prevailing wind is from the east, with the nearest major upwind city, Belem, approximately 1250 km away. The surrounding tropical forest emits vast quantities of biogenic gases and aerosol. Few roads connect Manaus to the rest of Brazil, and most freight and traffic from outside the city are via ship or plane. Thus, Manaus acts as a large point source of anthropogenic emissions which are transported to the surrounding Amazon Basin. As part of this campaign, the DOE Gulfstream 1 (G-1) research aircraft conducted two 6-week-long missions in which it investigated the evolution of the Manaus plume as it was transported into the surrounding Amazon tropical rain forest. The timing of these flight missions was chosen to provide a contrast between the wet and dry season (Martin et al., 2016). In the wet season, back-trajectory analysis indicated that the Manaus region was typically under the influence of air originating from the North Atlantic Ocean (Martin et al., 2016). Regular, organized mesoscale convective systems triggered by sea breeze circulation brought widespread, moderate-rate precipitation to the region (Giangrande et al., 2017; Machado et al., 2018; Burleyson et al., 2016). The high level of rainfall and moisture in the wet season inhibited biomass burning, and a low number of fires were observed (Martin et al., 2016; Machado et al., 2018). Under these conditions, the Amazon Basin is one of the cleanest continental regions on Earth, onto which the Manaus plume represents a significant perturbation (Martin et al., 2010b; Artaxo et al., 2013). In the dry season, back-trajectory analysis indicates that air masses originate from the Southern Hemisphere and travel up the Amazon River, transporting pollution from the northern coastal cities (Martin et al., 2016). In addition, recirculation events transported air from the southern Amazon Basin into the Manaus region (Martin et al., 2016). Intense biomass burning fires in the central and southern portion of the Amazon Basin and in central Africa were observed in the dry season, and a portion of these emissions were transported to the Manaus region (Martin et al., 2016, 2010b; Artaxo et al., 2013). In the dry season, more intense but less frequent convection produced approximately one quarter of the total rainfall observed in the wet season (Machado et al., 2018). As a result of the combination of transport and precipitation frequency, the Manaus region is significantly more polluted in the dry season.
In this paper we report on measurements from instruments deployed on the G-1, focusing primarily on measurements of aerosol species and trace gases that impact the aerosol life cycle. In the first part of the paper, we provide an overview of aerosol and volatile organic carbon (VOC) measurements and compare and contrast the wet- and dry-season data. In the second portion of the paper, we examine, in detail, the first 4–6.5 h of photochemical aging of the Manaus plume as it is transported into the surrounding tropical forest and interacts with biogenic emissions.
Table 1Flight times, altitudes of level legs, and meteorological parameters measured by instrumentation on board the G-1 aircraft for research flights described in this paper. Temperature, RH, wind speed, and wind heading correspond to values measured shortly after takeoff at altitudes less than 1000 m. Note that meteorological parameters are not homogeneous in space or time. Date format: month/day/year.
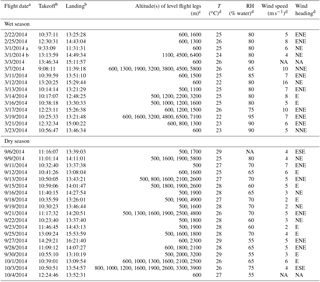
a The flight on 10 March 2014 is omitted from this
table due to an AMS failure. b Local time. c Altitude
above mean sea level from GPS data; only level flight legs on which AMS
data were collected are listed. d Immediately after takeoff, altitude
<1000 m, wind speed and heading from Aventech Research AIMMS-20 probe.
T from Rosemount 102E probe, RH calculated from T, and dew point measured
by General Eastern 1011-B probe.
2.1 G-1 flight strategy
The GoAmazon 2014/5 campaign was conducted in the vicinity of Manaus, Brazil, from 1 January 2014 through 31 December 2015 (Martin et al., 2016). During this period, DOE's G-1 research aircraft, based out of the Manaus International Airport, was deployed for two periods, during which time it sampled the Manaus urban plume as it was transported downwind over the Amazon rain forest. The first aircraft deployment period occurred during the wet season from 15 February to 26 March 2014, while the second occurred in the dry season from 1 September to 10 October 2014. Sixteen research flights were conducted in the wet season, and 19 were conducted in the dry season. Table 1 lists takeoff and landing times, the altitude of level flight legs, and meteorological parameters measured by the G-1 instrumentation shortly after takeoff on each flight. In general, flight plans were focused on successive perpendicular plume crossings spaced approximately equally at intervals of 24 km downwind of the city. A representative flight path for 13 March 2014, an exemplary day in the wet season with few clouds, is shown in Fig. 1. The paths for all flights during the two deployments are shown, segregated by altitude, in Martin et al. (2016) and therefore are not reproduced here. On 13 March, the first leg approximately bisected the city along a NW–SE line, the second leg passed near a highly instrumented ground site directly across the Amazon River from Manaus (T2), and the fourth leg passed over a second highly instrumented site located NE of Manacapuru (T3), with an additional leg between the T2 and T3 sites, and a final leg downwind of the T3 site. The first pass through the pattern was generally at an altitude of 500–700 m above ground level, with a second pass at a higher altitude and often multiple altitudes and often, though not always, focused on sampling clouds. The flight pattern discussed above and shown in Fig. 1 was rotated to align with the prevailing wind direction. Flights generally departed in the late morning or early afternoon and lasted between 3 and 3.5 h (Table 1).
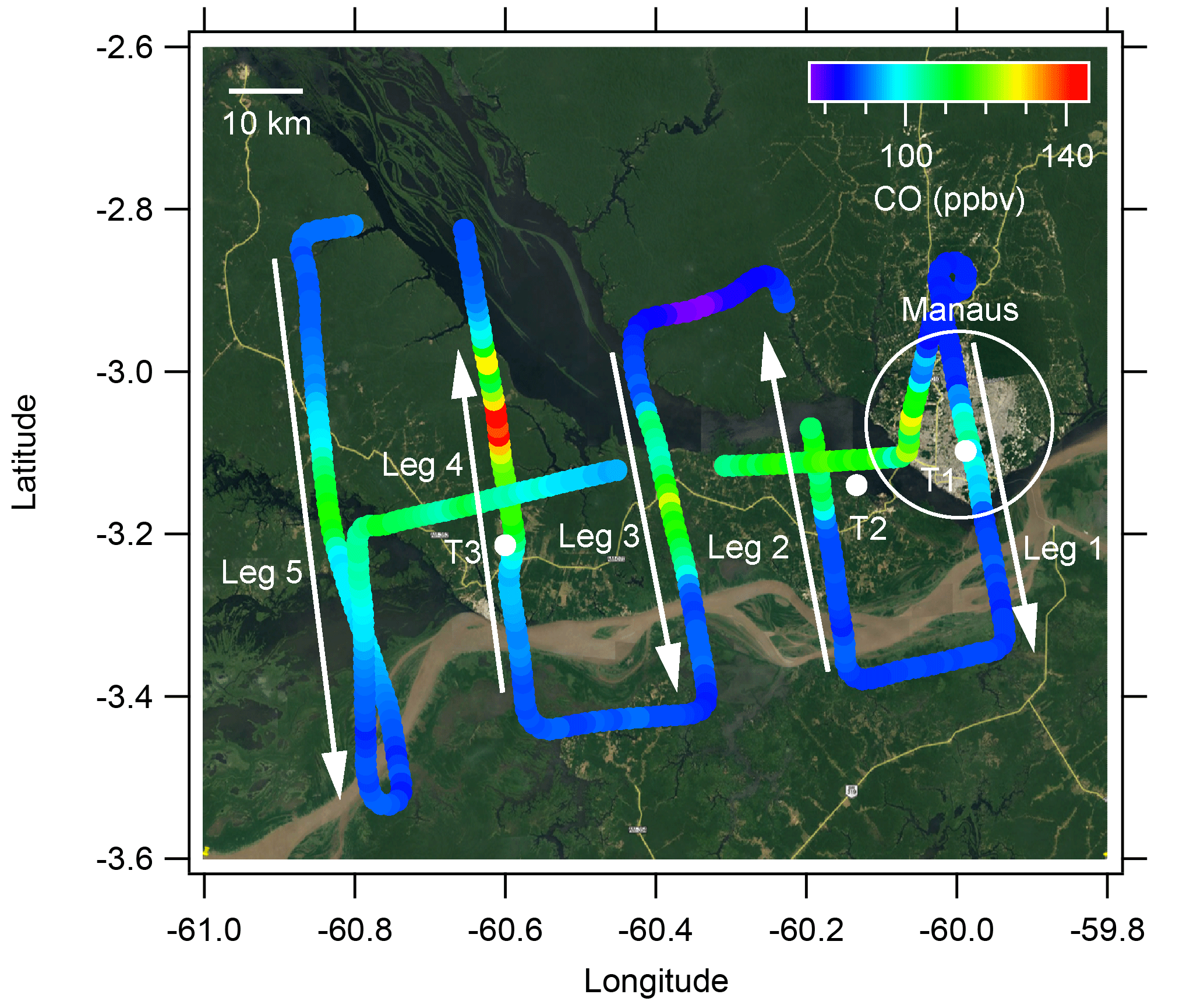
Figure 1Flight path for the 13 March 2014 flight colored by the CO concentration in units of ppm. Arrows indicate the direction of the flight. The first pass through the pattern was conducted at 500 m and then repeated at 1000 m. The G-1 took off at 10:12 LT and landed at 13:21 LT for a total flight duration of 3 h and 9 min. The white dots show the location of ground measurement sites. The underlying image is from Google Earth.
2.2 Instrumentation
An Aerodyne High-Resolution Time-of-Flight Aerosol Mass Spectrometer (abbreviated as AMS hereafter) was deployed on the G-1 to measure aerosol chemical composition (Jayne et al., 2000; DeCarlo et al., 2006). The AMS operated only in the standard “V” mass spectrometer (MS) mode (the particle sizing mode was not used) with a 13 s data averaging interval and equal chopper open and closed periods of 3 s. Before, during, and after flights, the AMS-sampled air was periodically diverted through a HEPA filter to remove particulates, and these filter periods were used to account for gas-phase interferences with isobaric particulate signals. Based on the standard deviation of these blank measurements (3σ) as described in the literature (DeCarlo et al., 2006), the detection limits of the AMS at the 13s sampling interval were 0.12, 0.01, 0.014, and 0.005 µg m−3 for organics, sulfate, nitrate, and ammonium, respectively. All aerosol instruments, including the AMS, sampled from a common, double-diffuser isokinetic inlet. Flow for the AMS was subsampled from the center of the isokinetic inlet and dried to RH <40 % by passage through a 1∕2 in. ID Perma Pure Nafion membrane. A constant-pressure inlet operating at approximately 620 hPa was used to maintain a constant volumetric flow to the AMS up to altitudes of approximately 3900 m (Bahreini et al., 2008). Because the AMS was powered off between flights, a correction based on the real-time signal was applied to all data to account for drifting sensitivity as the instrument warmed up during flights. The AMS was regularly calibrated in the field using monodisperse ammonium nitrate particles quantified with a TSI condensation particle counter (CPC). Data were analyzed in Igor Pro (v6.37) using the high-resolution analysis package (Squirrel v1.55H, PIKA v1.44H) and techniques described in the literature (Canagaratna et al., 2015; Kroll et al., 2011; Aiken et al., 2007; Allan et al., 2003; Jimenez et al., 2003). All AMS data discussed in this paper have been processed using the high-resolution data analysis routine. The O : C and H : C values reported here use the updated calibrations described in Canagaratna et al. (2015). All AMS data are normalized to the laboratory calibration conditions of 23 ∘C and 1013 hPa. Positive matrix factorization (PMF) analysis was performed on the wet-season data set by combining all flight data into a single experiment using the PMF Evaluation Tool (v 2.06) and the PFM2 algorithm (v 4.2) (Ulbrich et al., 2009; Paatero, 1997; Paatero and Tapper, 1994).
An Ionicon quadrupole high-sensitivity Proton-Transfer-Reaction Mass Spectrometer (PTR-MS) was used to measure selected gas-phase VOC concentrations (Lindinger et al., 1998). The PTR-MS was run in the ion monitoring mode in which signals of a limited number of pre-selected of m∕z values are sequentially measured, with one measurement cycle taking 3.5 s. Averaging time at each m∕z in the series varied depending on the sensitivity of the instrument to that species, the expected concentration, and the background, but it was generally between 0.2 and 0.5 s. Isoprene at m∕z 69 was sampled multiple times during the cycle to enable flux analysis (Gu et al., 2017). Drift tube temperature, pressure, and voltage were held at 60 ∘C, 2.22 hPa, and 600 V, respectively, resulting in an electric-field-to-gas-density (E∕N) ratio of 134 Td (1 Td cm2 V−1 s−1). The PTR-MS sampled air through a dedicated forward-facing inlet that consisted of approximately of 1∕4 in. OD stainless steel followed by approximately 46 in. of 1∕4 in. Teflon tubing and 36 in. of 1∕16 in. OD PEEK tubing. The flow through the Teflon tubing was 600 cc min−1 with 300 cc min−1 subsampled through the PEEK tubing for introduction into the PTR-MS. To assess the PTR-MS background, air was periodically diverted through a stainless-steel tube filled with a Shimadzu platinum catalyst heated to 600 ∘C, which removes VOCs from the airstream without perturbing RH. The catalyst efficiency was tested during the campaign by comparing signal from air containing VOCs passed through the catalyst with signal from VOC-free air. The PTR-MS was calibrated by introducing known concentrations of calibration gases into the instrument with variable dilution by VOC-free air. The calibration tank VOC concentrations were determined gravimetrically and verified using gas chromatography analysis by the manufacturer (AiR Environmental, Inc).
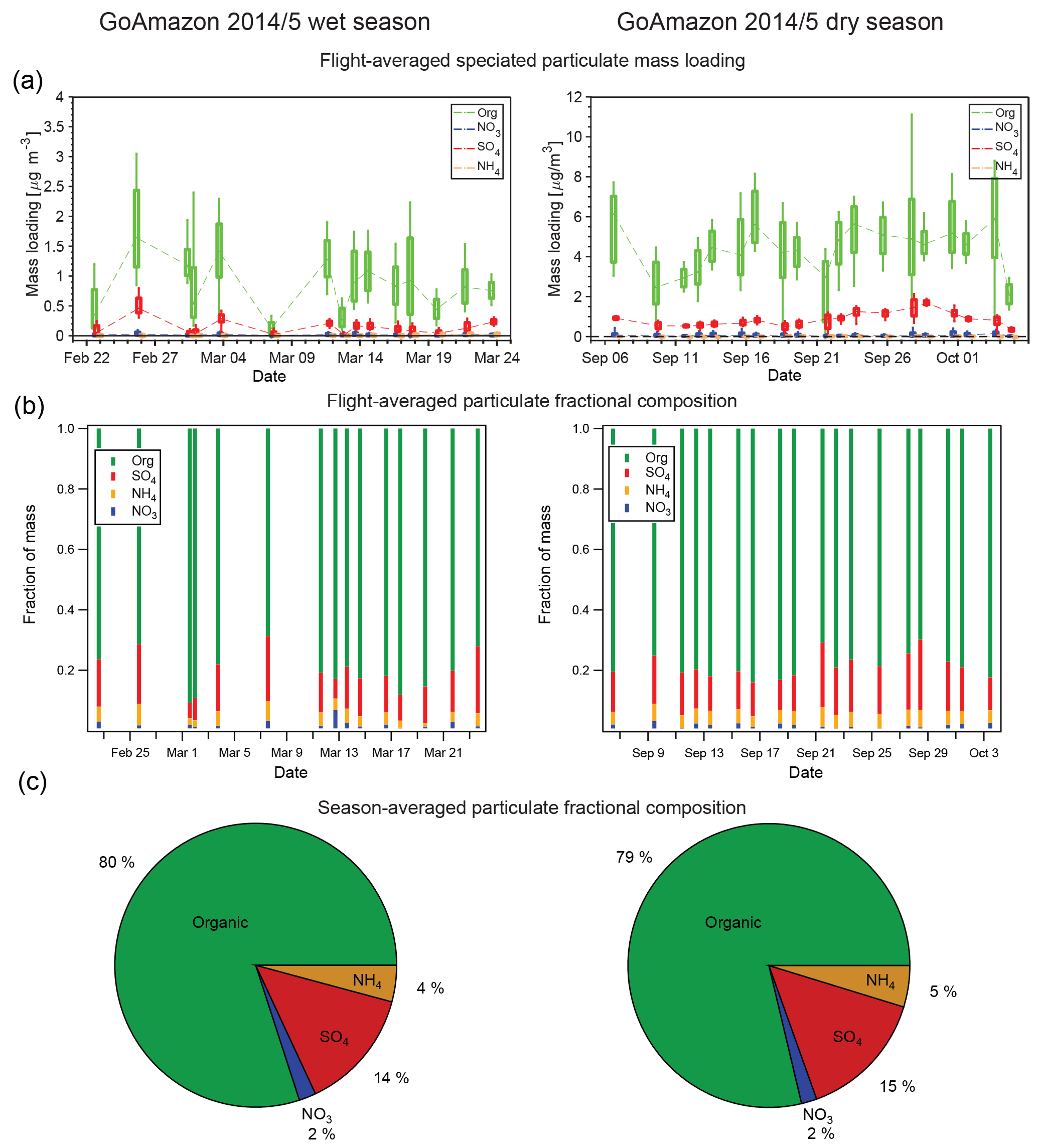
Figure 2Box-and-whisker representations of the G-1 AMS data for both the wet and dry seasons (a), average particle chemical composition for each flight (b), and average aerosol chemical composition for all flights (c). Boxes represent the quartiles, with whiskers extending to 10 % and 90 % and the median values shown as dots. Lines between the boxes connect the median and are drawn to guide the eye. Note the scale change between the wet- and dry-season box-and-whisker plots. AMS data are normalized to 23 ∘C and 1013 hPa.
Ozone was measured with a Thermo Scientific Model 49i ozone analyzer based on measurement of UV absorption at 254 nm. The instrument was regularly calibrated in flight by displacement of known quantities of ozone and zeroed in flight using ozone-scrubbed ambient air. CO was measured using a Los Gatos Research CO–N2O–H2O analyzer that is based on cavity-enhanced near-IR absorption and was also calibrated regularly in flight. Additional information on the instrumentation deployed on the G-1 can be found in the GoAmazon 2014/5 overview (Martin et al., 2016).
3.1 Overview of G-1 aerosol data and comparison of wet and dry seasons
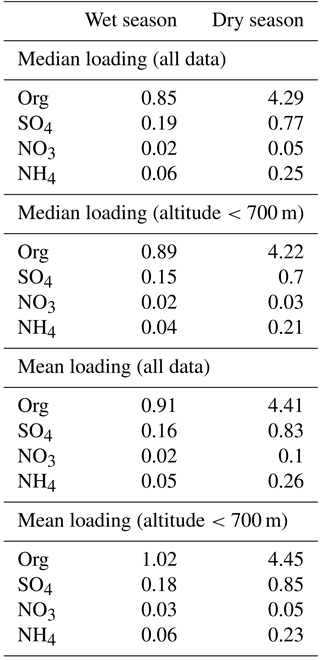
As discussed in the Introduction, the different precipitation and mesoscale transport patterns in the wet and the dry season are expected to produce considerably different concentrations of pollutants in the Amazon Basin, and measurements confirmed this expectation. Figure 2 and Table 2 summarize the AMS results for data collected on the G-1 flights in both the wet and the dry season. The top panel of Fig. 2 shows box-and-whisker plots summarizing AMS data for organic, sulfate, nitrate, and ammonium particulate concentrations, with one data point representing each flight (note the scale change between the wet- and dry-season plot). The middle panel shows the relative distribution of the chemical species on each flight. The bottom panel shows the distribution of the chemical species as a mass-weighted average of all data from the respective season. Several trends are readily apparent in the data. First, it is clear that aerosol loadings were significantly higher in the dry season than in the wet season. Median organic loadings for the wet- and dry-season data were 0.85 and 4.29 µg m−3, respectively. Median sulfate, nitrate, and ammonium loadings increased from 0.19, 0.02, and 0.06 µg m−3 in the wet season to 0.77, 0.05, and 0.25 µg m−3 in the dry season. Mean loadings of all species were somewhat higher than the median. Mean organic, sulfate, nitrate, and ammonium loadings were 0.91, 0.16, 0.02, and 0.05 µg m−3 in the wet season and 4.41, 0.80, 0.1, and 0.26 µg m−3 in the dry season. In the wet season, the mass loading of all species were lower than typically observed over continental regions in the Northern Hemisphere (Zhang et al., 2007).
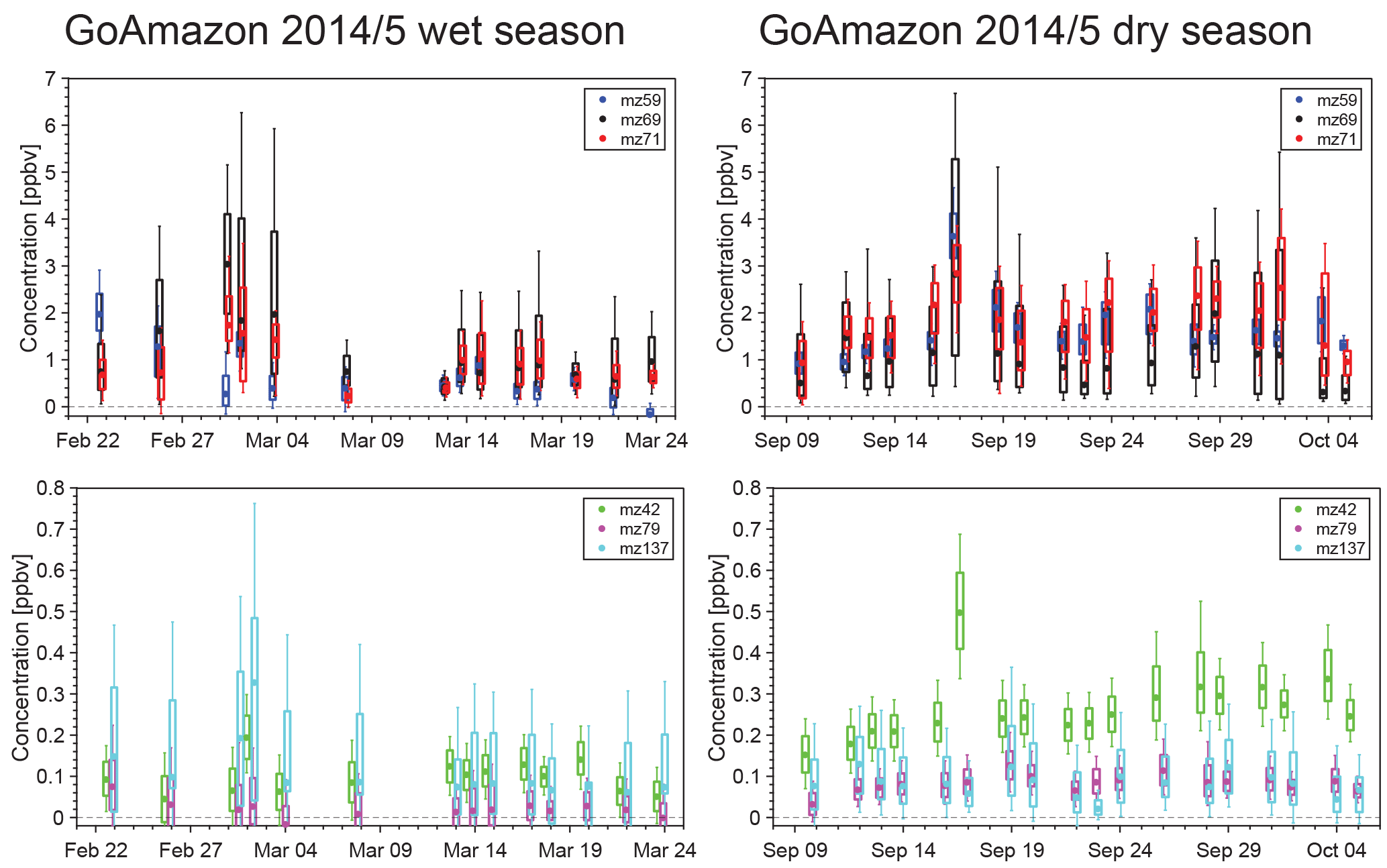
Figure 3Box-and-whisker representations of the G-1 PTR-MS data for both the wet and dry seasons. Boxes represent the quartiles, with whiskers extending to 10 % and 90 % and the median values shown as dots. Note the scale change between the top and bottom panels.
Table 2Chemically resolved mass loading measured by the AMS on board the G-1. Mass loadings are normalized to 23 ∘C and 1013 hPa. Units are µg m−3. The statistics for altitudes below 700 m approximate the boundary layer conditions at the time of the flights.
While this paper will focus on the particulate data, measurements of the volatile organic compounds provide insights into the precursors that are oxidized to form OA and help to identify the source of an air parcel. Figure 3 and Table 3 summarize the concentrations of several relevant VOCs measured on board the G-1 with the PTR-MS. Similar to the trends seen in the aerosol mass loading data, concentrations of most VOCs measured by the PTR-MS are significantly higher in the dry season than in the wet season. Concentrations of isoprene (m∕z 69) and its oxidation products (m∕z 71), biogenic precursors of OA, are a factor of 2–3 higher in the dry season than the wet season. The average daily high temperatures in Manaus are 33.5 ∘C in September (dry season) and 30.9 ∘C in March (wet season) (INMET, 2018), and isoprene emissions have been shown to scale with temperature, among other variables (Guenther et al., 2012). Measurements of benzene (m∕z 79), which is primarily anthropogenic in origin, were also significantly higher in the dry season. While benzene itself is unlikely to significantly contribute to SOA formation over the timescales we observe on the G-1 flights (4–6.5 h) due to its ∼5-day atmospheric lifetime (Atkinson and Arey, 2003), other unmeasured anthropogenic VOC concentrations which would contribute to more immediate OA formation may have been higher as well. The higher VOC concentrations may contribute to the higher OA concentrations measured in the dry season. However, the PTR-MS data, in addition to satellite and remote-sensing measurements (Martin et al., 2016), also show that biomass burning significantly impacted the region. Measurements of acetonitrile (m∕z 42), whose major source is biomass burning (Yokelson et al., 2009, 2007), are 2–3 times higher in the dry season than the wet season, indicating a significant biomass burning impact in the region. Biomass burning is a known source of OA (Jolleys et al., 2012; Bond et al., 2004; Yokelson et al., 2009; Ferek et al., 1998) and would also contribute to the higher aerosol concentrations observed in the dry season.
Table 3Measurements of VOC species made by the PTR-MS on board the G-1. Units are ppbv. The statistics for altitudes less than 700 m approximate the boundary layer conditions at the time of the flights.
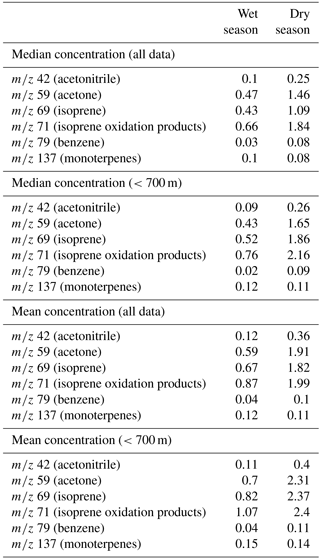
The aerosol mass loadings we measure in the dry season are consistent with aircraft-based AMS measurements made during the SAMBBA campaign at the same time of year, though several hundred kilometers to the SW in the state of Rondônia (Allan et al., 2014). Chen et al. (2009) reported AMS-measured wet-season campaign (7 February to 13 March 2008) average organic and sulfate loadings of 0.7 and 0.15 µg m−3 from a ground site that was within the GoAmazon 2014/5 flight domain, though they excluded data impacted by Manaus emissions from their analysis. When comparing aircraft to ground site data, it is important to acknowledge differences in the data sets. First, the aircraft samples a different spatial domain in all three spatial dimensions (latitude, longitude, and altitude) than a ground site. In addition, the G-1 typically sampled the boundary layer in the late morning or early afternoon (see Table 1 for flight times) and thus may have missed peak concentrations of secondary species. Finally, the goal of the flights was often to sample the Manaus plume for the first half of each flight and clouds for the second half. The mean aerosol loadings reported here agree well with those reported by Chen et al. (2009), considering the inherent differences in data sets discussed above. We also found good agreement when comparing the G-1-measured aerosol loadings to those measured at the T3 site when the aircraft passed overhead and at 500–600 m altitude (de Sá et al., 2018). Our loadings are at the low end of the range (1–2 µg m−3) reported from a ground site in Rondônia, approximately 825 km to the southwest of Manaus, though those data were more heavily influenced by biomass burning and relied on a combination of offline techniques to speciate the aerosol (Fuzzi et al., 2007). To understand potential bias in the data set due to altitude, we calculated statistics for data collected below 700 m, which captures the lowest altitude portion of each flight. Giangrande et al. (2017) analyzed radiosonde data from balloons launched at the T3 site and report average mixed-layer heights of greater than 1000 m above ground level at 10:00 local time in both the wet and dry seasons of GoAmazon 2014/5. Thus, the G-1 was typically sampling in the boundary layer on the lowest flight legs, and the data collected below 700 m should be representative of boundary layer conditions. In the wet season, the low-altitude AMS loading statistics are generally either slightly higher or unchanged relative to the full data set. In the dry season, the low-altitude AMS loading statistics are either slightly lower relative or unchanged relative to the full data set, likely due to transport of biomass burning from the south in elevated layers. Both the median and mean concentrations of most VOCs measured are slightly higher for the data at <700 m altitude relative to the full data set (Table 3). This is the expected behavior for most VOCs measured here as they either are directly emitted at the surface (e.g., isoprene, benzene, monoterpenes) or form from oxidation of VOCs emitted at the surface (e.g., isoprene oxidation products).
The fractional contribution of each species to the total loading is nearly identical when comparing seasons, despite the large differences in aerosol absolute mass concentrations and the larger influence of biomass burning in the dry season (Martin et al., 2016). Organics dominate the chemical composition, comprising 80 % of the total, with sulfate comprising 14 %, nitrate 2 %, and ammonium 4 %, regardless of the season. The relative distribution of the products changes only modestly among flights (Fig. 2, middle panel). Organics were the largest fraction of the total aerosol mass on each flight, varying between 69 and 90 %. Sulfate was the second-largest fraction of the total mass (6–20 %), and the sum of sulfate and organic was nearly constant. The fractional contributions of each chemical species described herein are nearly identical to the previous reports from Chen et al. (2009, 2015). The aircraft-measured chemical composition is also nearly identical to that measured at the T3 ground site (de Sá et al., 2018) during GoAmazon 2014/5. The agreement between the T3 and G-1 product distributions suggests that the T3 ground site is sampling air that is representative of the regional air and is not unduly biased by local emissions, at least in the aggregate.
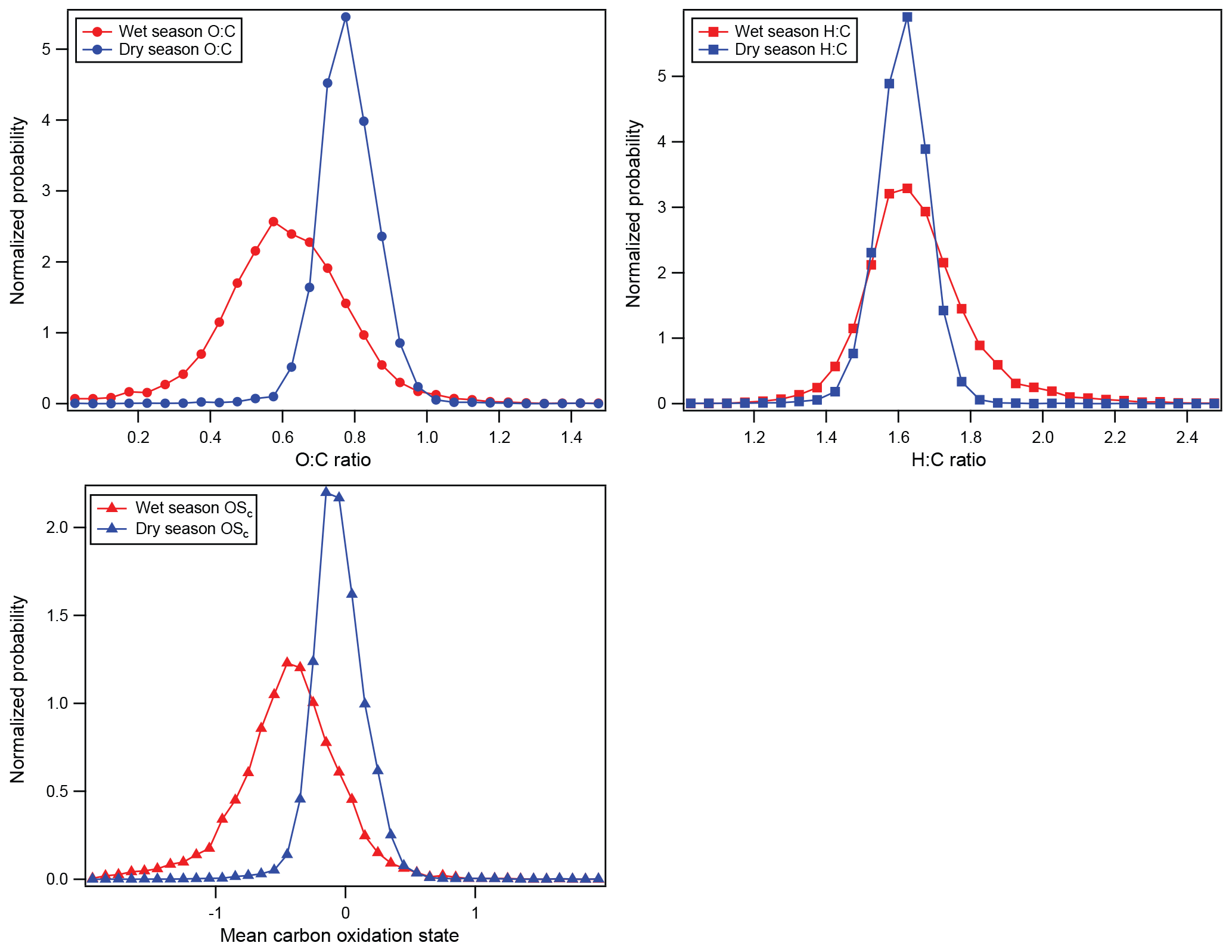
Figure 4Normalized occurrence probability calculated from G-1 HR-AMS analysis of the organic aerosol during the wet (red) and dry (blue) seasons. O : C and H : C were binned into 0.05-unit-wide bins, and data were binned in 0.1-unit-wide bins. O : C and H : C ratios were calculated using the updated methodology in Canagaratna et al. (2015). Mean carbon oxidation state is calculated according to Kroll et al. (2011).
The observation that the total concentrations of all aerosol components significantly increase yet remain in the same proportions when comparing the wet and the dry season is unexpected. The more frequent and widespread precipitation events in the wet season and the lower concentrations of precursor VOCs contribute to the lower aerosol concentrations in the wet season. If the aerosol particles were internally mixed, it would be reasonable to assume that rainfall would remove aerosol in approximately equal proportions. However, as discussed previously, aerosol sources are different between seasons, and there was a much larger biomass burning influence in the dry season (Martin et al., 2016), which is a known and significant source of OA (e.g., Jolleys et al., 2012; Bond et al., 2004; Yokelson et al., 2009; Ferek et al., 1998). Biomass burning was previously shown to emit particulate sulfate and nitrate directly, as well as NOx, sulfuric acid, and methane sulfonic acid (MSA), precursors of particulate nitrate and sulfate, but not in the same ratio as OA and its precursors (Yokelson et al., 2009). Thus, given the significant influence of biomass burning in the dry season, it is surprising that the chemical composition of the aerosol does not change.
Despite the similarity of the distribution of organic, sulfate, and nitrate in the aerosol particles between the wet and dry seasons, the chemical composition of the organic aerosol is quite different between seasons. Figure 4 shows normalized probability distributions for three metrics of the organic aerosol composition – the hydrogen-to-carbon ratio (H : C), the oxygen-to-carbon ratio (O : C), and the mean carbon oxidation state () – segregated by season. It is clear that the organic particles are more oxidized in the dry season than the wet season. The median O : C, H : C, and in the dry season were 0.78, 1.58, and −0.02, respectively, while they were 0.6, 1.65, and −0.45 in the wet season. While the H : C ratios change only slightly between season, the O : C and both show significant increases in the dry season. Thus, the aerosol in the dry season is significantly more aged, consistent with aged biomass burning aerosol contributing to the organic aerosol mass in the dry season and relatively fresh, locally generated organic aerosol dominating in the wet season (Martin et al., 2010b). The probability distributions during the wet season are also wider than in the dry season. We postulate that small contributions from a wider range of sources were able to influence the data to a larger degree in the wet season, when the total organic aerosol concentrations were much smaller. In contrast, during the dry season, small sources of organic mass would have a smaller impact on the organic composition due to the significant biomass burning background.
3.2 Evolution of organic aerosol in the Manaus plume
Figure 1 shows the flight path on 13 March 2014, which is an exemplary day for observing the evolution of the Manaus plume in the wet season. Flight legs were perpendicular to the prevailing wind direction and spaced approximately 24 km apart at successively increasing distance from Manaus. The length of the legs was chosen such that measurements (e.g., CO concentration) returned to near background levels at the ends of the leg. Due to dispersion of the plume, the length of the legs generally increased downwind of Manaus, ranging from 56 km close to the city to 80 km downwind of the T3 site. The pattern was flown first at an altitude of 500 m, with a second pass at 1000 m overlapping the first. This flight was marked by mostly sunny skies, with no clouds intercepted by the G-1 at 500 m and only brief passage through spatially small clouds at 1000 m. There was little local or regional biomass burning during this time period. Thus, this flight represents a case with coherent transport of the Manaus plume downwind into regions of high biogenic emissions with few complicating factors, such as biomass burning or cloud processing.
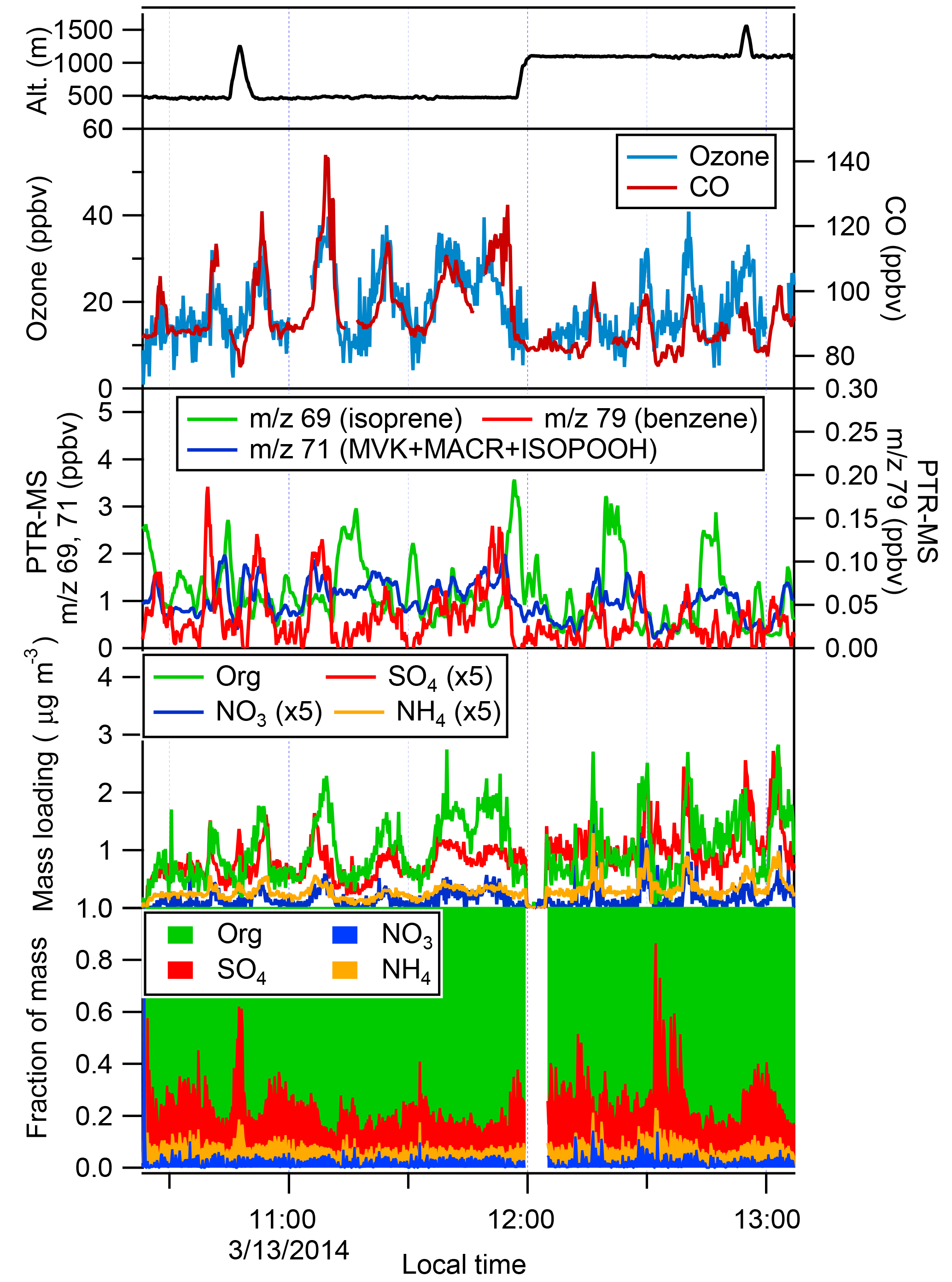
Figure 5Time traces of relevant plume measurements on the 13 March 2014 flight. Note the mass of inorganic species (SO4, NO3, NH4) has been multiplied by a factor of 5 to improve the figure clarity. PTR-MS signal at m∕z 69 corresponds to isoprene; m∕z 71 corresponds to the sum of methyl vinyl ketone (MVK), methacrolein (MACR), and isoprene hydroxyhydroperoxide (ISOPOOH), all oxidation products of isoprene; and m∕z 79 corresponds to benzene. AMS data are normalized to 23 ∘C and 1013 hPa.
Figure 5 shows a time series of several important gas- and particle-phase species for the 13 March 2014 flight. Passage through the Manaus plume is indicated by clear and significant increases in CO, ozone, and PTR-MS m∕z 79 (benzene) above the background levels. While CO may be produced from the oxidation of biogenic VOCs (BVOCs; Slowik et al., 2010), the intense peaks are primarily from urban emissions, given the proximity to Manaus and the correlation of CO with other known anthropogenic species, such as ozone, benzene, and toluene (not shown). Concentrations of CO (80 ppbv) and ozone (10 ppbv) are lowest at the edges of the flight legs; although we did not sample extensively in regions that were far removed from Manaus, these concentrations are consistent with the background concentrations that have been previously reported in the region (Andreae et al., 2015; Martin et al., 2010b; Harriss et al., 1990). Isoprene concentrations (PTR-MS m∕z 69) typically reach their highest levels outside of the plume and are lower inside, though isoprene concentrations of approximately 1 ppbv are still observed in the plume. As seen in Fig. 1, much of the land cover outside of Manaus is tropical forest, with some pasture land interspersed. Significant isoprene emissions are expected and indeed measured from the forest (Gu et al., 2017; Guenther et al., 2006). Significant concentrations of methacrolein, methyl vinyl ketone, and isoprene hydroxyhydroperoxides (PTR-MS m∕z 71), all first-generation oxidation products of isoprene, are also observed and have a more complicated structure (Liu et al., 2016). Concentrations of these products tend to be highest near the edges of the plume, and they do not correlate strongly with either isoprene (r2=0.24) or anthropogenic plume markers such as CO (r2=0.24). Production of these oxidation products requires OH, the concentration of which depends on NOx, to oxidize isoprene, and thus they are expected to have a more complex relationship with the urban plume and the biogenic emissions from the forest. Taken together, the observations of both isoprene and its oxidation products suggest that photooxidation of BVOCs is enhanced in the plume relative to the background, as would be expected (Martin et al., 2017). Monoterpene concentrations (not shown) on this flight are approximately a factor of 10 lower than isoprene concentrations, consistent with previous measurements (Kesselmeier et al., 2000), and are often near or below the instrument limit of detection (∼200 pptv). Monoterpene concentrations correlate with isoprene concentrations during the flight, when they are above the instrument detection limit.
OA mass increases from background values of 0.5 µg m−3 to as high as 2.8 µg m−3 in the Manaus plume, and on this day it is correlated relatively well with both ozone (r2=0.65) and CO (r2=0.76). While OA concentrations do not reach the levels that are typically seen in the outflow of large North American cities, they are significantly larger than background concentrations (up to 6×), indicating a clear impact of the plume on OA concentrations. Sulfate also clearly increase in the plume, though its peak concentrations occur toward the southern edge of the plume, relative to organics, CO, and ozone, suggesting sulfur emissions may not be co-located with other anthropogenic emissions. However, there are also occasional peaks in the sulfate concentration (for example at 10:47 LT) that coincide with minima in other plume indicators, such as CO and ozone. Nitrate concentrations are significantly lower than sulfate concentrations but tend to correlate more strongly with organic concentrations than sulfate concentrations. There are indications that a significant portion of the nitrate mass exists as organic nitrates, consistent with measurements at the T3 site and with aircraft measurements (Allan et al., 2014; de Sá et al., 2018; Schulz et al., 2018). The NO+ : ion ratio is 5–7 in the plume, compared to a NO+ : ion ratio of 1.2–1.5 measured during the campaign for ammonium nitrate calibration aerosol. NO+ : ion ratios significantly above that of ammonium nitrate are indicative of the presence of organic nitrates, though quantifying the organic fraction is challenging (Farmer et al., 2010). Ammonium concentrations tend to closely mirror sulfate concentrations. The bottom panel of Fig. 5 shows the fractional composition of the aerosol, which helps to illustrate the relative differences among the aerosol species. Organic species clearly dominate at all points, with sulfate the next-most-important species.
At 1000 m altitude, the plume can still be observed in the G-1 data, though the concentrations of plume markers such as CO, ozone, and benzene are lower. Increases in organic concentration are less sharp and defined than observed at 500 m, though enhancements are still clear. The organic mass fraction is somewhat smaller at 1000 m, and sulfate and ammonium mass fractions are larger. Based on radiosonde measurements, the boundary layer grew from 900 to 1200 m between the beginning and end of the flight on this date. Data from the G-1 suggest that the 1000 m flight path was near the top of the boundary layer. Thus, at 1000 m altitude, the data are influenced both by the local conditions and from air that has been transported over longer distances.
Figure 6 shows a PMF analysis of the organic species for the 13 March flight along with independently measured tracers that might be expected to correlate with the PMF factors. We were able to resolve the organic aerosol into three factors: an OOA factor that is a proxy for SOA; an HOA factor that is a proxy for primary organic particulate emissions; and an IEPOX SOA factor, which forms from the heterogeneous uptake of IEPOX, a product of isoprene oxidation under low-NOx conditions (Hu et al., 2015; Robinson et al., 2011; Lin et al., 2012; Zhang et al., 2011;, 2005a; de Sá et al., 2017). Details of the PMF analysis, including mass spectra of the factors, can be found in the Appendix and Supplement. As expected, both the OOA and HOA factor mass loadings are significantly higher in the plume relative to the background. The HOA factor correlates strongly with CO (r=0.79 for all data at 500 m), while the OOA factor correlates strongly with ozone (r=0.81 for all data at 500 m), though we will show below that the slope of the correlation changes with plume age. The changing slope has the effect of degrading the HOA–CO and OOA–ozone correlations, which are higher for individual plume legs. The HOA factor is a larger fraction of the organic mass on the legs nearest to Manaus and becomes a smaller fraction on the downwind legs. The OOA mass fraction displays the opposite behavior, increasing downwind as the plume ages. The transformation of HOA to SOA and/or OOA has been observed previously, both in laboratory experiments and in the field (Sage et al., 2008; Robinson et al., 2007; DeCarlo et al., 2008; Zhang et al., 2007; Ng et al., 2010). Taken together, these observations are consistent with the literature assignment of HOA as a marker of primary organic emissions and OOA as a marker of secondary organic aerosol. Outside of the plume, HOA concentrations approach zero. OOA concentrations, on the other hand, remain significant and approach the organic aerosol background concentrations. The IEPOX SOA factor does not appear to change with passage through the plume and does not correlate with sulfate aerosol on this day as may be expected based on the chemical mechanism, which requires heterogeneous uptake of gas-phase IEPOX onto inorganic aerosol to generate IEPOX SOA (Hu et al., 2015; Surratt et al., 2010; Gaston et al., 2014; Nguyen et al., 2014). We note that the IEPOX SOA factor is noisy, is near or below the limit of detection of organic aerosol (0.13 µg m−3) for much of this flight, and is shown largely for the sake of completeness. Because of the low concentrations, it is difficult to draw definitive conclusions about IEPOX SOA formation on this flight. However, given the focus of this flight was on tracking the Manaus plume, which contains significant NOx, our findings are consistent with de Sá et al. (2017), who report that NOx from the Manaus plume suppresses IEPOX SOA formation in the plume and that the IEPOX SOA factor was less than 5 % of the total OA at T3 at the time of the G-1 overflight.

Figure 6PMF analysis of the organic aerosol on the 13 March 2014 flight. AMS data are normalized to 23 ∘C and 1013 hPa.
Figure 7 summarizes several relevant plume quantities at 500 m altitude as a function of the approximate distance downwind from Manaus, which serves as a proxy for the photochemical age of the air mass. The ratio of excess organic aerosol to excess CO (Δorg∕ΔCO) is a metric that is often used to quantify organic aerosol formation in a source plume as it ages. The utility of the Δorg∕ΔCO metric rests mainly on the assumptions that CO is conserved on the timescale of the measurements and that urban emissions scale linearly with CO. Our calculation also assumes that Manaus emissions are the dominant source of CO in the plume. Using this ratio rather than absolute concentrations of organic aerosol can normalize for dilution due to mixing of the plume with background air. The Δorg∕ΔCO values shown in Fig. 7 were calculated as the slope of a linear regression line between the AMS-measured organic mass loadings and CO concentrations. All data for each flight leg perpendicular to the wind direction were included. Background values of OA and CO were not subtracted from the data, and the regression was not forced through the origin. We acknowledge that calculation of Δorg∕ΔCO values can be sensitive to the calculation method and choice of background values for both OA and CO; therefore, we performed calculations using different methods and assuming different background concentrations for both OA and CO and find general agreement between these methods (see Supplement for more details).
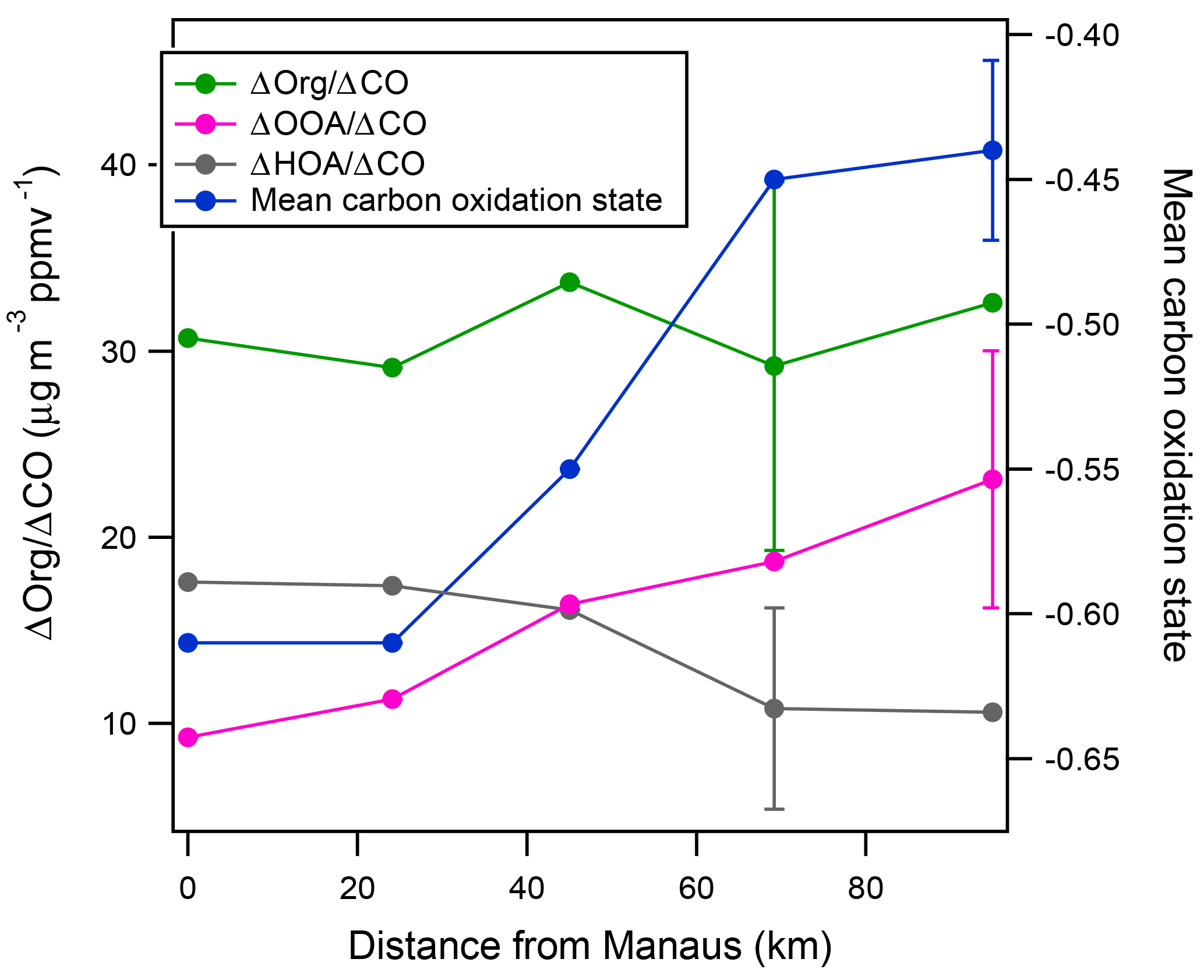
Figure 7Key metrics describing the evolution of the Manaus urban plume on 13 March. Each data point represents the average values for one pass through the plume. A representative error bar is shown for each set of measurements. Mean wind speeds were 7.3 m s−1 on this flight, thus data capture approximately the first 4–5 h of the plume aging. Calculations of ΔOrg∕ΔCO values use method one with details on the calculations and methods provided in the text and the Supplement. Spatial locations of each leg are labelled in Fig. 1.
Two aspects of the Δorg∕ΔCO distinguish these measurements from most previous measurements in the literature. First, Δorg∕ΔCO is ∼31 µg m−3 ppmv−1, a value that is lower than the value of 70±20 µg m−3 ppmv−1 observed in the outflow of cities in North America (e.g., de Gouw and Jimenez, 2009, and references therein). The Δorg∕ΔCO values observed here are also significantly smaller than the values of 77–157 µg m−3 ppmv−1 we previously observed when fresh urban anthropogenic emissions evolved in the presence of strong biogenic emissions, as is the case in the present data set (Shilling et al., 2013; Setyan et al., 2012). We measure a ΔHOA∕ΔCO value of 17.6 µg m−3 ppmv−1 near Manaus (Fig. 7), which should represent fresh emissions. This value is at the upper end of what is reported in the literature for North American cities, indicating that lower primary particulate emissions from Manaus are not responsible for the lower Δorg∕ΔCO values (DeCarlo et al., 2010; de Gouw and Jimenez, 2009).
The second new aspect of these observations is that we do not observe a corresponding increase in Δorg∕ΔCO as the plume photochemically ages, contrary to our expectations. We note that, as described above, organic aerosol concentrations are significantly higher in the plume than in the background, and the constant Δorg∕ΔCO values do not indicate that no OA formation occurs in the plume. Rather, the Δorg∕ΔCO analysis that follows focuses on understanding the aging of the Manaus plume. Based on literature observations (de Gouw and Jimenez, 2009; Kleinman et al., 2008; DeCarlo et al., 2010; Takegawa et al., 2006; Sullivan et al., 2006; Freney et al., 2014), we hypothesized that Δorg∕ΔCO would rapidly increase in the Manaus plume due to OA formation from sources not associated with urban CO emissions (Setyan et al., 2012; Shilling et al., 2013). Specifically, we expected that enhanced OH concentrations in the plume would lead to rapid oxidation of BVOCs emitted from the surrounding forest, which would produce OA with little concomitant production of CO and lead to a rapid increase in Δorg∕ΔCO. As seen in Fig. 7, the data did not support this hypothesis. As described in the Supplement, we performed the Δorg∕ΔCO calculation using several methods and assuming different background CO and OA values and did not find significant increases in Δorg∕ΔCO in any of these calculations. Furthermore, though we focus on Δorg values based on the AMS data, we also calculated Δvolume∕ΔCO using aerosol size distribution data from two independent instruments, the Ultra-High Sensitivity Aerosol Spectrometer (UHSAS) and the Fast Integrated Mobility Spectrometer (FIMS), that were also on board the G-1. Neither of these calculations indicates an increase in Δvolume∕ΔCO with plume age.
Previous studies have observed increases in Δorg∕ΔCO with plume age, with the largest increases occurring for the first ∼1 day of aging and changes in Δorg∕ΔCO gradually levelling off beyond approximately 1–2 days (DeCarlo et al., 2010; de Gouw and Jimenez, 2009; Kleinman et al., 2008; Freney et al., 2014). Based on the mean wind speeds observed along the flight track (7.3 m s−1) and the transport distance (up to 100 km), we estimate that the plume was 4–5 h old at the farthest leg and freshly emitted over the city. Unfortunately, photochemical clocks could not be used to more precisely calculate the photochemical age of the plume (de Gouw et al., 2005; Kleinman et al., 2003; Parrish et al., 1992). NOy measurements were not available on this flight. Benzene and toluene concentrations were low and noisy, particularly at increasingly downwind distances from Manaus. Though our observations are limited to shorter aging timescales (4–5 h) than many literature studies (1–2 days), the literature studies report measurable changes in Δorg∕ΔCO at short aging timescales (Kleinman et al., 2008; DeCarlo et al., 2010). Furthermore, we clearly observe other indicators of photochemical aging in the plume. Ozone concentrations are 30–50 ppbv in the plume, compared to background levels of 10–15 ppbv, indicating active photochemistry. Concentrations of isoprene are lower in the plume than the background values observed outside of it. Concentrations of isoprene inside the plume do not show a monotonic dependence on plume age. The concentration of isoprene oxidation products measured in the plume by the PTR-MS at m∕z 71 monotonically increases with plume aging, from 0.96 ppbv directly over the city to 1.27 ppbv at the farthest leg. The average toluene : benzene ratio in the plume also monotonically decreases with plume aging as would be expected, though attempts to convert these ratios into a photochemical age resulted in unrealistically high estimates of the plume age, likely due to noise in the data as mentioned above (de Gouw et al., 2005). The mean particle oxidation state () of plume OA increases from −0.6 to −0.44 as it ages. It is expected that photochemical aging would produce progressively more oxygenated species downwind of Manaus that subsequently partition to the aerosol phase, increasing the mean carbon oxidation state. Other studies have observed a similar phenomenon as particles were transported downwind of urban centers (DeCarlo et al., 2010). The particles size distributions measured by both the FIMS and the UHSAS also indicate that particles size increases downwind of Manaus. As discussed in the previous section, we also observe that the mass fraction of HOA decreases and simultaneously the mass fraction of OOA increases as the plume ages. As seen in Fig. 7, ΔHOA∕ΔCO is 17.6 µg m−3 ppmv−1 on the leg nearest to the city and decreases to 10.6 µg m−3 ppmv−1. At the same time, ΔOOA∕ΔCO increases from 9.2 to 23.1 µg m−3 ppmv−1. Thus, some fraction of HOA appears to be lost – through volatilization, deposition, or a combination of both – with the lost HOA mass balanced by an increase in OOA mass. All of these factors indicate that active photochemistry is occurring and is transforming VOCs into SOA and aging the particles. Thus, we would expect to see significant increases in Δorg∕ΔCO for these aging times if similar observations of outflow from North American cities were representative of the Manaus plume.
The sum of the HOA and OOA factors explains >95 % of the total OA mass within the plume, as the IEPOX SOA mass is small. Though calculating the ΔHOA∕ΔCO and ΔOOA∕ΔCO ratios introduces some noise (particularly ΔOOA∕ΔCO close to Manaus), the sum of these ratios is approximately constant with aging and equal to the Δorg∕ΔCO values. Thus, the total OA mass is conserved with loss and volatilization of HOA balanced out by formation of OOA. This process appears to explain the lack of an increase in Δorg∕ΔCO with aging. It is difficult to determine whether the OOA carbon mass is anthropogenic in origin, biogenic, or (most likely) a combination of both. Volatilized HOA and co-emitted anthropogenic VOCs would undergo oxidation in the plume, and the products may condense and be detected as OOA (Sage et al., 2008; Robinson et al., 2007). In addition, we see evidence that biogenic VOCs are oxidized in the plume, likely more rapidly than in the background due to enhanced in-plume OH concentrations, and these oxidation products may also condense as OOA. Either mechanism or a combination of both would explain the observed increase in the carbon oxidation state. It is surprising that the volatilized HOA mass is compensated by oxidation and recondensation of SOA for these relatively fresh particles.
Thus far, we have focused on a detailed analysis of the 13 March 2014 plume because there is a clear contrast between the background and plume data through the portion of the flight in the boundary layer, there is little interference from biomass burning and cloud processing, the flight pattern was extended far enough downwind to investigate a significant aging time, and the data from all key instruments are available and complete. In an effort to understand whether the Δorg∕ΔCO observations on 13 March are representative of plume aging in the wet season, we have screened the rest of the wet-season flight data and, based on the criteria mentioned above, identified two additional flights, 11 and 16 March 2014, for analysis. The Δorg∕ΔCO values for these data as a function of distance from Manaus are shown in Fig. 8. On 11 March, Δorg∕ΔCO values average 29.9 µg m−3 ppmv−1, show little change with distance from Manaus, and show similar variability between legs to the 13 March flight data. The mean wind speed for the analyzed portion of the flight was 5.3 m s−1, translating to an approximate maximum plume age of 5–5.5 h. On 16 March, Δorg∕ΔCO values average 42.0 µg m−3 ppmv−1, show little significant increase with distance from Manaus, and exhibit a larger range of values than the other two data sets. The mean wind speed for the analyzed portion of the 16 March flight was 4.6 m s−1, translating to an approximate maximum plume age of 6–6.5 h. We estimate that the error in the Δorg∕ΔCO measurement is approximately 10 µg m−3 ppmv−1; thus the difference in average Δorg∕ΔCO values between the 16 March and the 11 and 13 March data sets is at the edge of what we consider significant. The average Δorg∕ΔCO is 34.3 µg m−3 ppmv−1 for all flights, and the combined data set collectively represents the first 4–6.5 h of aging. In addition to the aircraft data, Cirino et al. (2018) report that the median Δorg∕ΔCO values for the plume as measured at the T2 and T3 sites are nearly identical to one another in the wet season and similar to measurements from the G-1 (Cirino et al., 2018). Thus, the data from both G-1 and the ground sites suggest that Δorg∕ΔCO values of 30–40 µg m−3 ppmv−1, which change little in the first 4–6.5 h of plume aging, are representative of the Manaus plume behavior in the wet season. As previously discussed, most studies examining outflow of North American cities have shown changes in Δorg∕ΔCO that are largest at the shortest aging times, and we are unaware of reports of constant Δorg∕ΔCO in an aging urban plume. Both the identity and the distribution of Manaus's emissions may very well be distinct from those of a typical North American city. In addition, background concentrations of anthropogenic pollutants are lower and biogenic VOC emissions are higher in the unperturbed rain forest surrounding Manaus relative to most North American cities. Finally, background OA concentrations near Manaus are lower in the wet season than in most other continental regions, and SOA yield is known to increase with OA mass loading (Odum et al., 1996). All of these factors may contribute of the difference in the Δorg∕ΔCO observations.
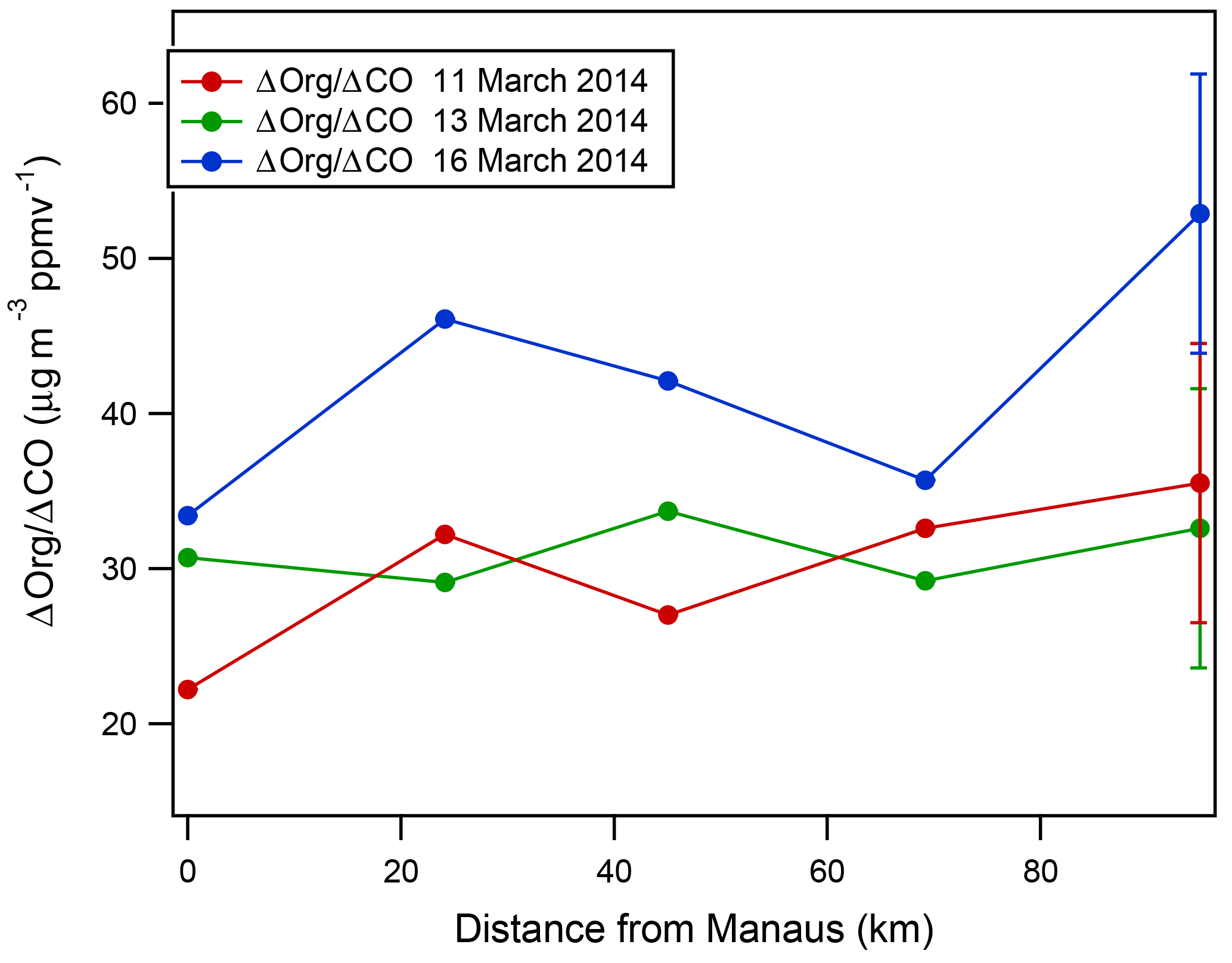
Figure 8Δorg∕ΔCO measurements in the plume as a function of distance from Manaus for 11, 13, and 16 March 2014. A representative error bar is shown for each set of measurements. Mean wind speeds were 5.8, 7.3, and 4.6 m s−1 on 11, 13, and 16 March, respectively. Collectively, the data capture approximately the first 4–6.5 h of the plume aging.
A similar phenomenon, clear chemical aging with little change of Δorg∕ΔCO, has been observed in field studies of biomass burning plumes by several researchers (Jolleys et al., 2012; Cubison et al., 2011; Hecobian et al., 2011; Akagi et al., 2012; Forrister et al., 2015). Clearly, the mix of organic compounds emitted from a forest fire and from the Manaus urban region will be significantly different, and this is not to imply that the detailed chemical mechanisms are the same. However, the similarity of the observations is suggestive that a similar process could occur in both types of plumes.
3.3 Sources of sulfate in the Manaus region
Sulfate was the second-largest contributor to the non-refractory aerosol mass during the GoAmazon 2014/5 campaign (accounting for 13 % of the PM1 mass in both the wet and dry season), so it is also instructive to examine the source of sulfate in the area. In 2014 Manaus generated much of its power from fuel oil and diesel, which are known to emit SO2 that eventually oxidizes to form condensable sulfate (Medeiros et al., 2017). However, several observations suggest that the major source of sulfate in the plume may not come from local SO2 emissions. First, on many flights, particulate sulfate levels clearly increases in the plume (e.g., Fig. 5), including on the legs closest to Manaus and thus very near the source. The lifetime of SO2 with respect to homogeneous oxidation by OH is expected to be about 1 week, and only a small fraction (∼5 % on the farthest leg) of SO2 emissions would have time to oxidize to H2SO4 in the gas phase. In addition, there is no indication of dramatically enhanced sulfate loading downwind of Manaus on the 13 March flight, when the ΔSO4∕ΔCO ratio averages 2.6 µg m−3 ppmv−1 and does not increase with plume age. On the 13 March flight, there was no precipitation and there were relatively few clouds along the flight track, so heterogeneous oxidation of sulfate in cloud water cannot explain the sulfate enhancement seen in the plume.
We can possibly attribute these observations to two mechanisms. First, it is possible that natural sulfur emissions originating downwind of Manaus undergo oxidation by enhanced concentrations of OH in the Manaus plume and subsequently condense (Andreae et al., 1990; Chen et al., 2009). Andreae et al. (1990) estimate that in-basin sulfur emissions are responsible for approximately 0.05 µg m−3 of the total sulfate loading. Measurements during the AMAZE-08 campaign also suggested natural in-basin emissions and transport of natural out-of-basin emission were a significant source of particulate sulfate (Chen et al., 2009). A 0.05 µg m−3 sulfate loading is roughly consistent with our measured sulfate concentrations outside of the plume and would account for 15–30 % of the sulfate in the plume on the 13 March flight. Higher OH concentrations in the plume would speed oxidation of natural sulfur compounds and further increase the fraction of in-plume sulfate originating from natural emissions. Second, power plant plumes may directly emit sulfuric acid, which then may condenses on pre-existing particles. A previous aircraft study of the Manaus outflow observed high concentrations of particles smaller than 40 nm in power plant plumes, which they attribute to sulfuric acid (Kuhn et al., 2010). Though particles in this size range are unlikely to be detected by the AMS, condensation of sulfuric acid on larger, pre-existing particles would also contribute to the in-plume sulfate observations.
In summary, we report on measurements of the aerosol chemical composition, sources, and evolution of the urban plume from research flights conducted in both the wet and dry season in the vicinity of Manaus, Brazil. Median mass loading of organics, sulfate, nitrate, and ammonium was 0.85, 0.19, 0.02, and 0.06 µg m−3 in the wet season and 4.29, 0.77, 0.05, and 0.25 µg m−3 in the dry season, respectively. Despite the significant difference in mass loadings, the average fractional composition of the aerosol did not change significantly between seasons. Organics dominate the aerosol chemical composition in both seasons, comprising 80 % of the total non-refractory aerosol mass. The OA was significantly more oxidized in the dry season with a of −0.02 in the dry season and −0.45 in the wet season.
The flight on 13 March 2014 was a golden day to study the evolution of the Manaus plume as it advected to the surrounding Amazon tropical forest. The organic and sulfate aerosol concentrations were both significantly enhanced in the plume along with CO, ozone, and benzene. The spatial distribution of the sulfate aerosol was shifted toward the southern edge of the plume, relative to organics, CO, and benzene. As the plume is transported downwind of Manaus, we observe a change in the relative fraction of HOA and OOA mass. ΔHOA∕ΔCO values decreased from 17.6 µg m−3 ppmv−1 over Manaus to 10.6 µg m−3 ppmv−1 95 km downwind of Manaus, indicating a substantial loss of HOA mass. This loss of HOA mass was balanced out by OOA formation, with ΔOOA∕ΔCO values increasing from 9.2 to 23.1 µg m−3 ppmv−1 during the 4–5 h aging timescale. Concomitantly, the mean carbon oxidation state of the OA increased from −0.6 to −0.44. Because the loss of HOA mass was balanced out by addition of OOA mass, net changes in Δorg∕ΔCO with plume age were not observed and averaged 31 µg m−3 ppmv−1. Analysis of data from two additional fights during the wet season also found no net changes in Δorg∕ΔCO with plume age. The average Δorg∕ΔCO for all the analyzed G-1 data was 34 µg m−3 ppmv−1 and collectively represents the first 4–6.5 h of plume aging. The observation of constant Δorg∕ΔCO with aging was in contrast to our hypothesis that Δorg∕ΔCO would increase rapidly due to plume-enhanced oxidation of BVOCs, emitted by the surrounding tropical forest and not associated with CO emissions, and subsequent conversion to OA mass. Our Δorg∕ΔCO observations are also in contrast to literature observations of the outflow of several different North American urban centers, which have shown increases in Δorg∕ΔCO for the first 1–2 days of plume aging (Kleinman et al., 2008; de Gouw and Jimenez, 2009; DeCarlo et al., 2010; Freney et al., 2014; Sullivan et al., 2006).
The data set generated from the G-1 measurements during GoAmazon 2014/5 provide a set of observations for understanding aerosol chemistry in the Amazon region. Our preliminary analysis shows that aging of the Manaus plume generates less OA in the wet season when compared to the aging of the outflow of many North American cities. The differences are likely due to a combination of factor including differences in emissions from both Manaus and the surrounding tropical forest, lower levels of background anthropogenic pollution in the Amazon, and lower background OA concentrations into which semi-volatile organics can partition. These results have implications for modeling efforts and for understanding how urban pollution impacts the surrounding pristine Amazon.
All ARM data sets used in this study may be downloaded from the ARM website at https://www.arm.gov/research/campaigns/amf2014goamazon (last access: 24 May 2018).
Positive matrix factorization (PMF) analysis was performed on the high-resolution organic aerosol mass spectra and error matrix that were measured using the HR-ToF-AMS on the G-1. The organic aerosol mass spectra () and error matrix were initially prepared using the high-resolution AMS analysis package (Squirrel v1.55H, PIKA v1.44H) as described in the literature (Jimenez et al., 2003; Allan et al., 2004; DeCarlo et al., 2006; Aiken et al., 2007, 2008). The AMS organic mass loading data were normalized to the laboratory calibration conditions of 23 ∘C and 1013 hPa. Error was estimated from the ion-counting statistics (Allan et al., 2003), and a minimum error was assigned to each signal (Ulbrich et al., 2009). Ions with signal below zero (due to noise) were removed from the matrix. In addition, ions with an average signal-to-noise ratio (S∕N) less than 0.75 were removed, and ions with average S∕N between 0.75 and 2 were downweighted by a factor of 2 (Ulbrich et al., 2009). Ions whose signals are assigned based on the measured CO2 signal in the ratios described by Aiken et al. (2008) (CO+, H2O+, HO+, O+) are also downweighted by a factor of 2 (Ulbrich et al., 2009). The PFM2 algorithm version 4.2 (Paatero, 1997; Paatero and Tapper, 1994) was used to analyze the matrix, and results were visualized and evaluated using the PMF Evaluation Tool (v 2.06) described by Ulbrich et al. (2009). PMF analysis of this data set was challenging, due the combination of low concentrations of organic aerosol, fast sampling required for aircraft operations, and the often small changes in aerosol concentrations across the flight domain. PMF analysis on data from a single flight often (though not always) resulted in a failure to resolve factors that are described in the literature (e.g., HOA, IEPOX SOA). Instead, we often observes split factors that are largely dominated by a single high-S∕N ion (CHO+, , C2H3O+). For example, the IEPOX SOA factor was not resolved when the 13 March flight was analyzed on its own, largely due to the small mass loading of this factor (Fig. 6). Therefore, data from all flights during the wet season were combined into a single matrix and analyzed together, which also forces a common solution for the entire data set.
Table A1Summary of scaled residuals and reasoning used in the choice of a five-factor solution in the PMF analysis of the wet-season HR-AMS data.

We chose a five-factor solution to the combined matrix based on a number of criteria, including the residuals and fit quality, the correlation of chosen factors with independently measured species (e.g., CO, ozone), the realism of the factor mass spectra (are the spectra representative of a physically realistic aerosol component?), and the comparison of the factor spectra to literature data. Table A1 summarizes our decision-making and lists the residuals as a function of the number of PMF factors. As seen in Table A1, the residuals continue to significantly decrease in moving from a two- to five-factor solution. For example, Q∕Qexp decreases from 0.28 to 0.24, and the fraction of the residual aerosol mass decreases from 4 % to 0.1 % in moving from the two to the five-factor solution. Figure S2 in the Supplement shows the residuals and scaled residuals for each time point in the matrix for the five-factor solution, indicating that all the data are well fit. Solutions with three or fewer factors failed to separate the HOA and the IEPOX SOA factors, which are well described in the literature and were reasonably expected to be present in the data. One of the OOA-like spectra in the three-factor solution also appeared to have signs of both the HOA and IEPOX SOA factors, as evidenced by significant signal of the key marker ions (m∕z 57) for HOA and C5H6O+ (m∕z 82) for IEPOX SOA. In the four-factor solution, an HOA factor was resolved along with three OOA-like spectra, though the OOA spectra show evidence of factor splitting (discussed below). The five-factor solution was able to resolve HOA, IEPOX SOA, and three OOA-like factors.
Figures S3 and S4 show the time series and mass spectra for the five-factor solution. Examination of the mass spectra (Fig. S4) suggests that the OOA factor has split. For example, the mass spectrum of factor 3 is dominated by the CHO+ ion with little signal through the rest of the spectra. The spectrum of factor 1 is dominate by and other ions that are based on the signal (CO+, H2O+, HO+, O+) in prescribed ratios (Aiken et al., 2008). These factors are unlikely to represent some real aerosol component, as no known OA produces such simple spectra. In addition, the time series of factors 1 and 2 are highly correlated in time (see Fig. S5). In summary, two factors are highly correlated in time (factors 1 and 2), and a third factor is dominated by a single ion. Thus, we recombine factors 1, 2, and 3 into a single factor, which we label OOA. The spectra of the resultant three factors (after recombining) are shown in Fig. S6, and the recombined factor is presented in the main text. The mathematical reason for the factor splitting is clear: aerosol loading is low, the AMS is sampling at a relatively fast rate (13 s averaging time), and the AMS spectra are dominated by a relatively small number of high-S∕N ions. In separate analyses, we downweighted the CHO+, C2H3O+, and ion signals by factors of 2–10 in an attempt to minimize the splitting of the OOA factor but were unsuccessful.
Comparison of the factor mass spectra to literature PMF spectra and correlation of the factor time profiles with independent measurements provides further support for the 5-factor solution. The combined OOA factor compares well with the spectra presented in the literature for OOA (e.g., Zhang et al., 2007, 2011, 2005b; Ng et al., 2010). As shown in Fig. 6, the OOA factor correlates well with ozone, a known secondary species that also forms through photochemical reactions. Similarly, the spectrum of factor 4 compares well with the mass spectrum of HOA widely reported in the literature (e.g., Zhang et al., 2007, 2011, 2005b; Ng et al., 2010). As discussed in the text and shown in Fig. 6, the HOA factor correlates well with CO, a known tracer of anthropogenic combustion processes. We identify factor 5 as the IEPOX SOA factor that is widely reported in the literature and believed to represent SOA formed from the heterogeneous uptake of isoprene epoxydiols onto pre-existing aerosol (Hu et al., 2015; Robinson et al., 2011; Lin et al., 2012). Our assignment is based on a comparison of the factor mass spectral profile with those in the literature, e.g., Hu et al. (2015).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-10773-2018-supplement.
JES, MSP, ECF, SdS, JMH, SRS, JT, and JW collected measurements and analyzed data. PA, KML, LATM, JW, and STM designed the field campaign and flight plans. JES prepared the manuscript with contributions from the co-authors.
The authors declare that they have no conflict of interest.
This article is part of the special issue “Observations and Modeling of the Green Ocean Amazon (GoAmazon2014/5) (ACP/AMT/GI/GMD inter-journal SI)”. It is not associated with a conference.
The authors thank the G-1 flight and ground crews for supporting the
GoAmazon 2014/5 mission. Funding for data collection on board the G-1
aircraft and at the ground sites was provided by the Atmospheric Radiation
Measurement (ARM) Climate Research Facility, a U.S. Department of Energy
(DOE) Office of Science user facility sponsored by the Office of Biological
and Environmental Research (OBER). Data analysis and research were supported
by the U.S. DOE's Atmospheric System Research Program under contract
DE-AC06-76RLO 1830 at PNNL. PNNL is operated for the U.S. DOE by Battelle
Memorial Institute. We acknowledge the support from the Central Office of
the Large Scale Biosphere Atmosphere Experiment in Amazonia (LBA), the
Instituto Nacional de Pesquisas da Amazonia (INPA), and the Instituto
Nacional de Pesquisas Espaciais (INPE). Paulo Artaxo acknowledges
Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP)
grants 2013/05014-0 and 2017/17047-0. The work was conducted under licenses
001262/2012-2 and 001030/2012-4 of the Brazilian National Council for
Scientific and Technological Development (CNPq).
Edited by: Tuukka Petäjä
Reviewed by: two anonymous referees
Aiken, A. C., DeCarlo, P. F., and Jimenez, J. L.: Elemental analysis of organic species with electron ionization high-resolution mass spectrometry, Anal. Chem., 79, 8350–8358, https://doi.org/10.1021/ac071150w, 2007.
Aiken, A. C., DeCarlo, P. F., Kroll, J. H., Worsnop, D. R., Huffman, J. A., Docherty, K., Ulbrich, I. M., Mohr, C., Kimmel, J. R., Sueper, D., Zhang, Q., Sun, Y., Trimborn, A., Northway, M., Ziemann, P. J., Canagaratna, M. R., Alfarra, R., Prevot, A. S. H., Dommen, J., Duplissy, J., Metzger, A., Baltensperger, U., and Jimenez, J. L.: O ∕ C and OM ∕ OC Ratios of Primary, Secondary, and Ambient Organic Aerosols with High Resolution Time-of-Flight Aerosol Mass Spectrometry, Environ. Sci. Technol., 42, 4478–4485, https://doi.org/10.1021/es703009q, 2008.
Akagi, S. K., Craven, J. S., Taylor, J. W., McMeeking, G. R., Yokelson, R. J., Burling, I. R., Urbanski, S. P., Wold, C. E., Seinfeld, J. H., Coe, H., Alvarado, M. J., and Weise, D. R.: Evolution of trace gases and particles emitted by a chaparral fire in California, Atmos. Chem. Phys., 12, 1397–1421, https://doi.org/10.5194/acp-12-1397-2012, 2012.
Allan, J. D., Jiminez, J. L., Williams, P. I., Alfarra, M. R., Bower, K. N., Jayne, J. T., Coe, H., and Worsnop, D. R.: Quantitative sampling using an Aerodyne aerosol mass spectrometer – 1. Techniques of data interpretation and error analysis, J. Geophys. Res., 108, 4090, https://doi.org/10.1029/2002JD002358, 2003.
Allan, J. D., Delia, A. E., Coe, H., Bower, K. N., Alfarra, M. R., Jimenez, J. L., Middlebrook, A. M., Drewnick, F., Onasch, T. B., Canagaratna, M. R., Jayne, J. T., and Worsnopf, D. R.: A generalised method for the extraction of chemically resolved mass spectra from aerodyne aerosol mass spectrometer data, J. Aerosol Sci., 35, 909–922, 2004.
Allan, J. D., Morgan, W. T., Darbyshire, E., Flynn, M. J., Williams, P. I., Oram, D. E., Artaxo, P., Brito, J., Lee, J. D., and Coe, H.: Airborne observations of IEPOX-derived isoprene SOA in the Amazon during SAMBBA, Atmos. Chem. Phys., 14, 11393–11407, https://doi.org/10.5194/acp-14-11393-2014, 2014.
Andreae, M. O.: Atmosphere. Aerosols before pollution, Science, 315, 50–51, https://doi.org/10.1126/science.1136529, 2007.
Andreae, M. O., Berresheim, H., Bingemer, H., Jacob, D. J., Lewis, B. L., Li, S. M., and Talbot, R. W.: The Atmospheric Sulfur Cycle over the Amazon Basin .2. Wet Season, J. Geophys. Res.-Atmos., 95, 16813–16824, https://doi.org/10.1029/JD095iD10p16813, 1990.
Andreae, M. O., Artaxo, P., Beck, V., Bela, M., Freitas, S., Gerbig, C., Longo, K., Munger, J. W., Wiedemann, K. T., and Wofsy, S. C.: Carbon monoxide and related trace gases and aerosols over the Amazon Basin during the wet and dry seasons, Atmos. Chem. Phys., 12, 6041–6065, https://doi.org/10.5194/acp-12-6041-2012, 2012.
Andreae, M. O., Acevedo, O. C., Araùjo, A., Artaxo, P., Barbosa, C. G. G., Barbosa, H. M. J., Brito, J., Carbone, S., Chi, X., Cintra, B. B. L., da Silva, N. F., Dias, N. L., Dias-Júnior, C. Q., Ditas, F., Ditz, R., Godoi, A. F. L., Godoi, R. H. M., Heimann, M., Hoffmann, T., Kesselmeier, J., Könemann, T., Krüger, M. L., Lavric, J. V., Manzi, A. O., Lopes, A. P., Martins, D. L., Mikhailov, E. F., Moran-Zuloaga, D., Nelson, B. W., Nölscher, A. C., Santos Nogueira, D., Piedade, M. T. F., Pöhlker, C., Pöschl, U., Quesada, C. A., Rizzo, L. V., Ro, C.-U., Ruckteschler, N., Sá, L. D. A., de Oliveira Sá, M., Sales, C. B., dos Santos, R. M. N., Saturno, J., Schöngart, J., Sörgel, M., de Souza, C. M., de Souza, R. A. F., Su, H., Targhetta, N., Tóta, J., Trebs, I., Trumbore, S., van Eijck, A., Walter, D., Wang, Z., Weber, B., Williams, J., Winderlich, J., Wittmann, F., Wolff, S., and Yáñez-Serrano, A. M.: The Amazon Tall Tower Observatory (ATTO): overview of pilot measurements on ecosystem ecology, meteorology, trace gases, and aerosols, Atmos. Chem. Phys., 15, 10723–10776, https://doi.org/10.5194/acp-15-10723-2015, 2015.
Artaxo, P., Rizzo, L. V., Brito, J. F., Barbosa, H. M. J., Arana, A., Sena, E. T., Cirino, G. G., Bastos, W., Martin, S. T., and Andreae, M. O.: Atmospheric aerosols in Amazonia and land use change: from natural biogenic to biomass burning conditions, Faraday Discuss., 165, 203–235, https://doi.org/10.1039/C3FD00052D, 2013.
Atkinson, R. and Arey, J.: Atmospheric Degradation of Volatile Organic Compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Bahreini, R., Dunlea, E. J., Matthew, B. M., Simons, C., Docherty, K. S., DeCarlo, P. F., Jimenez, J. L., Brock, C. A., and Middlebrook, A. M.: Design and operation of a pressure-controlled inlet for airborne sampling with an aerodynamic aerosol lens, Aerosol Sci. Technol., 42, 465–471, https://doi.org/10.1080/02786820802178514, 2008.
Bean, J. K., Faxon, C. B., Leong, Y. J., Wallace, H. W., Cevik, B. K., Ortiz, S., Canagaratna, M. R., Usenko, S., Sheesley, R. J., Griffin, R. J., and Hildebrandt Ruiz, L.: Composition and Sources of Particulate Matter Measured near Houston, TX: Anthropogenic-Biogenic Interactions, Atmosphere, 7, 23–73, https://doi.org/10.3390/atmos7050073, 2016.
Bond, T. C., Streets, D. G., Yarber, K. F., Nelson, S. M., Woo, J. H., and Klimont, Z.: A technology-based global inventory of black and organic carbon emissions from combustion, J. Geophys. Res.-Atmos., 109, D14203, https://doi.org/10.1029/2003jd003697, 2004.
Burleyson, C. D., Feng, Z., Hagos, S. M., Fast, J., Machado, L. A. T., and Martin, S. T.: Spatial Variability of the Background Diurnal Cycle of Deep Convection around the GoAmazon2014/5 Field Campaign Sites, J. Appl. Meteorol. Clim., 55, 1579–1598, https://doi.org/10.1175/jamc-d-15-0229.1, 2016.
Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R.: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 15, 253–272, https://doi.org/10.5194/acp-15-253-2015, 2015.
Chen, Q., Farmer, D. K., Schneider, J., Zorn, S. R., Heald, C. L., Karl, T. G., Guenther, A., Allan, J. D., Robinson, N., Coe, H., Kimmel, J. R., Pauliquevis, T., Borrmann, S., Poschl, U., Andreae, M. O., Artaxo, P., Jimenez, J. L., and Martin, S. T.: Mass spectral characterization of submicron biogenic organic particles in the Amazon Basin, Geophys. Res. Lett., 36, L20806, https://doi.org/10.1029/2009gl039880, 2009.
Chen, Q., Farmer, D. K., Rizzo, L. V., Pauliquevis, T., Kuwata, M., Karl, T. G., Guenther, A., Allan, J. D., Coe, H., Andreae, M. O., Pöschl, U., Jimenez, J. L., Artaxo, P., and Martin, S. T.: Submicron particle mass concentrations and sources in the Amazonian wet season (AMAZE-08), Atmos. Chem. Phys., 15, 3687–3701, https://doi.org/10.5194/acp-15-3687-2015, 2015.
Cirino, G. G., Brito, J., Barbosa, H. J. M., Rizzo, L. V., Carbone, S., de Sá, S. S., Tunved, P., Jimenez, J., Palm, B. B., Souza, R., Lavric, J., Tota, J., Oliveira, M., Wolff, S., Martin, S. T., and Artaxo, P.: Ground site observations of Manaus city plume evolution in GoAmazon2014/5, Atmos. Environ., submitted, 2018.
Crosier, J., Allan, J. D., Coe, H., Bower, K. N., Formenti, P., and Williams, P. I.: Chemical composition of summertime aerosol in the Po Valley (Italy), northern Adriatic and Black Sea, Q. J. Roy. Meteor. Soc., 133, 61–75, https://doi.org/10.1002/qj.88, 2007.
Cubison, M. J., Ortega, A. M., Hayes, P. L., Farmer, D. K., Day, D., Lechner, M. J., Brune, W. H., Apel, E., Diskin, G. S., Fisher, J. A., Fuelberg, H. E., Hecobian, A., Knapp, D. J., Mikoviny, T., Riemer, D., Sachse, G. W., Sessions, W., Weber, R. J., Weinheimer, A. J., Wisthaler, A., and Jimenez, J. L.: Effects of aging on organic aerosol from open biomass burning smoke in aircraft and laboratory studies, Atmos. Chem. Phys., 11, 12049–12064, https://doi.org/10.5194/acp-11-12049-2011, 2011.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
DeCarlo, P. F., Dunlea, E. J., Kimmel, J. R., Aiken, A. C., Sueper, D., Crounse, J., Wennberg, P. O., Emmons, L., Shinozuka, Y., Clarke, A., Zhou, J., Tomlinson, J., Collins, D. R., Knapp, D., Weinheimer, A. J., Montzka, D. D., Campos, T., and Jimenez, J. L.: Fast airborne aerosol size and chemistry measurements above Mexico City and Central Mexico during the MILAGRO campaign, Atmos. Chem. Phys., 8, 4027–4048, https://doi.org/10.5194/acp-8-4027-2008, 2008.
DeCarlo, P. F., Ulbrich, I. M., Crounse, J., de Foy, B., Dunlea, E. J., Aiken, A. C., Knapp, D., Weinheimer, A. J., Campos, T., Wennberg, P. O., and Jimenez, J. L.: Investigation of the sources and processing of organic aerosol over the Central Mexican Plateau from aircraft measurements during MILAGRO, Atmos. Chem. Phys., 10, 5257–5280, https://doi.org/10.5194/acp-10-5257-2010, 2010.
de Gouw, J. and Jimenez, J. L.: Organic aerosols in the Earth's atmosphere, Environ. Sci. Technol., 43, 7614–7618, https://doi.org/10.1021/es9006004, 2009.
de Gouw, J. A., Middlebrook, A. M., Warneke, C., Goldan, P. D., Kuster, W. C., Roberts, J. M., Fehsenfeld, F. C., Worsnop, D. R., Canagaratna, M. R., Pszenny, A. A. P., Keene, W. C., Marchewka, M., Bertman, S. B., and Bates, T. S.: Budget of organic carbon in a polluted atmosphere: Results from the New England Air Quality Study in 2002, J. Geophys. Res.-Atmos., 110, D16305, https://doi.org/10.1029/2004JD005623, 2005.
de Gouw, J. A., Brock, C. A., Atlas, E. L., Bates, T. S., Fehsenfeld, F. C., Goldan, P. D., Holloway, J. S., Kuster, W. C., Lerner, B. M., Matthew, B. M., Middlebrook, A. M., Onasch, T. B., Peltier, R. E., Quinn, P. K., Senff, C. J., Stohl, A., Sullivan, A. P., Trainer, M., Warneke, C., Weber, R. J., and Williams, E. J.: Sources of particulate matter in the northeastern United States in summer: 1. Direct emissions and secondary formation of organic matter in urban plumes, J. Geophys. Res., 113, D08301, https://doi.org/10.1029/2007jd009243, 2008.
de Sá, S. S., Palm, B. B., Campuzano-Jost, P., Day, D. A., Newburn, M. K., Hu, W., Isaacman-VanWertz, G., Yee, L. D., Thalman, R., Brito, J., Carbone, S., Artaxo, P., Goldstein, A. H., Manzi, A. O., Souza, R. A. F., Mei, F., Shilling, J. E., Springston, S. R., Wang, J., Surratt, J. D., Alexander, M. L., Jimenez, J. L., and Martin, S. T.: Influence of urban pollution on the production of organic particulate matter from isoprene epoxydiols in central Amazonia, Atmos. Chem. Phys., 17, 6611–6629, https://doi.org/10.5194/acp-17-6611-2017, 2017.
de Sá, S. S., Palm, B. B., Campuzano-Jost, P., Day, D. A., Hu, W., Isaacman-VanWertz, G., Yee, L. D., Brito, J., Carbone, S., Ribeiro, I. O., Cirino, G. G., Liu, Y. J., Thalman, R., Sedlacek, A., Funk, A., Schumacher, C., Shilling, J. E., Schneider, J., Artaxo, P., Goldstein, A. H., Souza, R. A. F., Wang, J., McKinney, K. A., Barbosa, H., Alexander, M. L., Jimenez, J. L., and Martin, S. T.: Urban influence on the concentration and composition of submicron particulate matter in central Amazonia, Atmos. Chem. Phys. Discuss., https://doi.org/10.5194/acp-2018-172, in review, 2018.
Ehn, M., Thornton, J. A., Kleist, E., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I. H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurten, T., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petaja, T., Wahner, A., Kerminen, V. M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Fan, J., Rosenfeld, D., Zhang, Y., Giangrande, S. E., Li, Z., Machado, L. A. T., Martin, S. T., Yang, Y., Wang, J., Artaxo, P., Barbosa, H. M. J., Braga, R. C., Comstock, J. M., Feng, Z., Gao, W., Gomes, H. B., Mei, F., Pöhlker, C., Pöhlker, M. L., Pöschl, U., and de Souza, R. A. F.: Substantial convection and precipitation enhancements by ultrafine aerosol particles, Science, 359, 411–418, https://doi.org/10.1126/science.aan8461, 2018.
Farmer, D. K., Matsunaga, A., Docherty, K. S., Surratt, J. D., Seinfeld, J. H., Ziemann, P. J., and Jimenez, J. L.: Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry, P. Natl. Acad. Sci. USA, 107, 6670–6675, https://doi.org/10.1073/pnas.0912340107, 2010.
Ferek, R. J., Reid, J. S., Hobbs, P. V., Blake, D. R., and Liousse, C.: Emission factors of hydrocarbons, halocarbons, trace gases and particles from biomass burning in Brazil, J. Geophys. Res.-Atmos., 103, 32107–32118, https://doi.org/10.1029/98jd00692, 1998.
Forrister, H., Liu, J., Scheuer, E., Dibb, J., Ziemba, L., Thornhill, K. L., Anderson, B., Diskin, G., Perring, A. E., Schwarz, J. P., Campuzano-Jost, P., Day, D. A., Palm, B. B., Jimenez, J. L., Nenes, A., and Weber, R. J.: Evolution of brown carbon in wildfire plumes, Geophys. Res. Lett., 42, 4623–4630, https://doi.org/10.1002/2015gl063897, 2015.
Freney, E. J., Sellegri, K., Canonaco, F., Colomb, A., Borbon, A., Michoud, V., Doussin, J.-F., Crumeyrolle, S., Amarouche, N., Pichon, J.-M., Bourianne, T., Gomes, L., Prevot, A. S. H., Beekmann, M., and Schwarzenböeck, A.: Characterizing the impact of urban emissions on regional aerosol particles: airborne measurements during the MEGAPOLI experiment, Atmos. Chem. Phys., 14, 1397–1412, https://doi.org/10.5194/acp-14-1397-2014, 2014.
Fuzzi, S., Decesari, S., Facchini, M. C., Cavalli, F., Emblico, L., Mircea, M., Andreae, M. O., Trebs, I., Hoffer, A., Guyon, P., Artaxo, P., Rizzo, L. V., Lara, L. L., Pauliquevis, T., Maenhaut, W., Raes, N., Chi, X., Mayol-Bracero, O. L., Soto-García, L. L., Claeys, M., Kourtchev, I., Rissler, J., Swietlicki, E., Tagliavini, E., Schkolnik, G., Falkovich, A. H., Rudich, Y., Fisch, G., and Gatti, L. V.: Overview of the inorganic and organic composition of size-segregated aerosol in Rondônia, Brazil, from the biomass-burning period to the onset of the wet season, J. Geophys. Res., 112, D01201, https://doi.org/10.1029/2005jd006741, 2007.
Gaston, C. J., Riedel, T. P., Zhang, Z., Gold, A., Surratt, J. D., and Thornton, J. A.: Reactive Uptake of an Isoprene-Derived Epoxydiol to Submicron Aerosol Particles, Environ. Sci. Technol., 48, 11178–11186, https://doi.org/10.1021/es5034266, 2014.
Giangrande, S. E., Feng, Z., Jensen, M. P., Comstock, J. M., Johnson, K. L., Toto, T., Wang, M., Burleyson, C., Bharadwaj, N., Mei, F., Machado, L. A. T., Manzi, A. O., Xie, S., Tang, S., Silva Dias, M. A. F., de Souza, R. A. F., Schumacher, C., and Martin, S. T.: Cloud characteristics, thermodynamic controls and radiative impacts during the Observations and Modeling of the Green Ocean Amazon (GoAmazon2014/5) experiment, Atmos. Chem. Phys., 17, 14519–14541, https://doi.org/10.5194/acp-17-14519-2017, 2017.
Glasius, M. and Goldstein, A. H.: Recent Discoveries and Future Challenges in Atmospheric Organic Chemistry, Environ. Sci. Technol., 50, 2754–2764, https://doi.org/10.1021/acs.est.5b05105, 2016.
Goldstein, A. H., Koven, C. D., Heald, C. L., and Fung, I. Y.: Biogenic carbon and anthropogenic pollutants combine to form a cooling haze over the southeastern United States, P. Natl. Acad. Sci. USA, 106, 8835–8840, https://doi.org/10.1073/pnas.0904128106, 2009.
Gu, D., Guenther, A. B., Shilling, J. E., Yu, H., Huang, M., Zhao, C., Yang, Q., Martin, S. T., Artaxo, P., Kim, S., Seco, R., Stavrakou, T., Longo, K. M., Tota, J., de Souza, R. A. F., Vega, O., Liu, Y., Shrivastava, M., Alves, E. G., Santos, F. C., Leng, G., and Hu, Z.: Airborne observations reveal elevational gradient in tropical forest isoprene emissions, Nat. Commun., 8, 15541, https://doi.org/10.1038/ncomms15541, 2017.
Guenther, A., Karl, T., Harley, P., Wiedinmyer, C., Palmer, P. I., and Geron, C.: Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature), Atmos. Chem. Phys., 6, 3181–3210, https://doi.org/10.5194/acp-6-3181-2006, 2006.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Harriss, R. C., Sachse, G. W., Hill, G. F., Wade, L. O., and Gregory, G. L.: Carbon-Monoxide over the Amazon Basin during the Wet Season, J. Geophys. Res.-Atmos., 95, 16927–16932, https://doi.org/10.1029/JD095iD10p16927, 1990.
Hecobian, A., Liu, Z., Hennigan, C. J., Huey, L. G., Jimenez, J. L., Cubison, M. J., Vay, S., Diskin, G. S., Sachse, G. W., Wisthaler, A., Mikoviny, T., Weinheimer, A. J., Liao, J., Knapp, D. J., Wennberg, P. O., Kürten, A., Crounse, J. D., Clair, J. St., Wang, Y., and Weber, R. J.: Comparison of chemical characteristics of 495 biomass burning plumes intercepted by the NASA DC-8 aircraft during the ARCTAS/CARB-2008 field campaign, Atmos. Chem. Phys., 11, 13325–13337, https://doi.org/10.5194/acp-11-13325-2011, 2011.
Hoyle, C. R., Boy, M., Donahue, N. M., Fry, J. L., Glasius, M., Guenther, A., Hallar, A. G., Huff Hartz, K., Petters, M. D., Petäjä, T., Rosenoern, T., and Sullivan, A. P.: A review of the anthropogenic influence on biogenic secondary organic aerosol, Atmos. Chem. Phys., 11, 321–343, https://doi.org/10.5194/acp-11-321-2011, 2011.
Hu, W. W., Campuzano-Jost, P., Palm, B. B., Day, D. A., Ortega, A. M., Hayes, P. L., Krechmer, J. E., Chen, Q., Kuwata, M., Liu, Y. J., de Sá, S. S., McKinney, K., Martin, S. T., Hu, M., Budisulistiorini, S. H., Riva, M., Surratt, J. D., St. Clair, J. M., Isaacman-Van Wertz, G., Yee, L. D., Goldstein, A. H., Carbone, S., Brito, J., Artaxo, P., de Gouw, J. A., Koss, A., Wisthaler, A., Mikoviny, T., Karl, T., Kaser, L., Jud, W., Hansel, A., Docherty, K. S., Alexander, M. L., Robinson, N. H., Coe, H., Allan, J. D., Canagaratna, M. R., Paulot, F., and Jimenez, J. L.: Characterization of a real-time tracer for isoprene epoxydiols-derived secondary organic aerosol (IEPOX-SOA) from aerosol mass spectrometer measurements, Atmos. Chem. Phys., 15, 11807–11833, https://doi.org/10.5194/acp-15-11807-2015, 2015.
INMET (Instituto Nacional de Meteorologia): Normais Climatologicas do Brasil 1981–2010, available at: http://www.inmet.gov.br/portal/index.php?r=clima/normaisClimatologicas, last access: 17 July 2018.
IPCC: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Stocker, T. F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S. K., Boschung, J., Nauels, A., Xia, Y., Bex, V., and Midgley, P. M., Cambridge University Press, Cambridge, UK and New York, NY, USA, 1535 pp., https://doi.org/10.1017/CBO9781107415324, 2013.
Jayne, J. T., Leard, D. C., Zhang, X. F., Davidovits, P., Smith, K. A., Kolb, C. E., and Worsnop, D. R.: Development of an aerosol mass spectrometer for size and composition analysis of submicron particles, Aerosol Sci. Technol., 33, 49–70, https://doi.org/10.1080/027868200410840, 2000.
Jimenez, J. L., Jayne, J. T., Shi, Q., Kolb, C. E., Worsnop, D. R., Yourshaw, I., Seinfeld, J. H., Flagan, R. C., Zhang, X. F., Smith, K. A., Morris, J. W., and Davidovits, P.: Ambient aerosol sampling using the Aerodyne Aerosol Mass Spectrometer, J. Geophys. Res.-Atmos., 108, 8425, https://doi.org/10.1029/2001jd001213, 2003.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Jolleys, M. D., Coe, H., McFiggans, G., Capes, G., Allan, J. D., Crosier, J., Williams, P. I., Allen, G., Bower, K. N., Jimenez, J. L., Russell, L. M., Grutter, M., and Baumgardner, D.: Characterizing the aging of biomass burning organic aerosol by use of mixing ratios: a meta-analysis of four regions, Environ. Sci. Technol., 46, 13093–13102, https://doi.org/10.1021/es302386v, 2012.
Kesselmeier, J., Kuhn, U., Wolf, A., Andreae, M. O., Ciccioli, P., Brancaleoni, E., Frattoni, M., Guenther, A., Greenberg, J., Vasconcellos, P. D., de Oliva, T., Tavares, T., and Artaxo, P.: Atmospheric volatile organic compounds (VOC) at a remote tropical forest site in central Amazonia, Atmos. Environ., 34, 4063–4072, https://doi.org/10.1016/S1352-2310(00)00186-2, 2000.
Kleinman, L. I., Daum, P. H., Lee, Y. N., Nunnermacker, L. J., Springston, S. R., Weinstein-Lloyd, J., Hyde, P., Doskey, P., Rudolph, J., Fast, J., and Berkowitz, C.: Photochemical age determinations in the Phoenix metropolitan area, J. Geophys. Res.-Atmos., 108, 4096, https://doi.org/10.1029/2002jd002621, 2003.
Kleinman, L. I., Springston, S. R., Daum, P. H., Lee, Y.-N., Nunnermacker, L. J., Senum, G. I., Wang, J., Weinstein-Lloyd, J., Alexander, M. L., Hubbe, J., Ortega, J., Canagaratna, M. R., and Jayne, J.: The time evolution of aerosol composition over the Mexico City plateau, Atmos. Chem. Phys., 8, 1559–1575, https://doi.org/10.5194/acp-8-1559-2008, 2008.
Kroll, J. H., Donahue, N. M., Jimenez, J. L., Kessler, S. H., Canagaratna, M. R., Wilson, K. R., Altieri, K. E., Mazzoleni, L. R., Wozniak, A. S., Bluhm, H., Mysak, E. R., Smith, J. D., Kolb, C. E., and Worsnop, D. R.: Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 3, 133–139, https://doi.org/10.1038/nchem.948, 2011.
Kuhn, U., Ganzeveld, L., Thielmann, A., Dindorf, T., Schebeske, G., Welling, M., Sciare, J., Roberts, G., Meixner, F. X., Kesselmeier, J., Lelieveld, J., Kolle, O., Ciccioli, P., Lloyd, J., Trentmann, J., Artaxo, P., and Andreae, M. O.: Impact of Manaus City on the Amazon Green Ocean atmosphere: ozone production, precursor sensitivity and aerosol load, Atmos. Chem. Phys., 10, 9251–9282, https://doi.org/10.5194/acp-10-9251-2010, 2010.
Lin, Y. H., Zhang, Z., Docherty, K. S., Zhang, H., Budisulistiorini, S. H., Rubitschun, C. L., Shaw, S. L., Knipping, E. M., Edgerton, E. S., Kleindienst, T. E., Gold, A., and Surratt, J. D.: Isoprene epoxydiols as precursors to secondary organic aerosol formation: acid-catalyzed reactive uptake studies with authentic compounds, Environ. Sci. Technol., 46, 250–258, https://doi.org/10.1021/es202554c, 2012.
Lindinger, W., Hansel, A., and Jordan, A.: On-line monitoring of volatile organic compounds at pptv levels by means of proton-transfer-reaction mass spectrometry (PTR-MS) – Medical applications, food control and environmental research, Int. J. Mass Spectrom., 173, 191–241, https://doi.org/10.1016/S0168-1176(97)00281-4, 1998.
Liu, Y., Brito, J., Dorris, M. R., Rivera-Rios, J. C., Seco, R., Bates, K. H., Artaxo, P., Duvoisin, S., Jr., Keutsch, F. N., Kim, S., Goldstein, A. H., Guenther, A. B., Manzi, A. O., Souza, R. A., Springston, S. R., Watson, T. B., McKinney, K. A., and Martin, S. T.: Isoprene photochemistry over the Amazon rainforest, P. Natl. Acad. Sci. USA, 113, 6125–6130, https://doi.org/10.1073/pnas.1524136113, 2016.
Machado, L. A. T., Calheiros, A. J. P., Biscaro, T., Giangrande, S., Silva Dias, M. A. F., Cecchini, M. A., Albrecht, R., Andreae, M. O., Araujo, W. F., Artaxo, P., Borrmann, S., Braga, R., Burleyson, C., Eichholz, C. W., Fan, J., Feng, Z., Fisch, G. F., Jensen, M. P., Martin, S. T., Pöschl, U., Pöhlker, C., Pöhlker, M. L., Ribaud, J.-F., Rosenfeld, D., Saraiva, J. M. B., Schumacher, C., Thalman, R., Walter, D., and Wendisch, M.: Overview: Precipitation characteristics and sensitivities to environmental conditions during GoAmazon2014/5 and ACRIDICON-CHUVA, Atmos. Chem. Phys., 18, 6461–6482, https://doi.org/10.5194/acp-18-6461-2018, 2018.
Martin, S. T., Andreae, M. O., Althausen, D., Artaxo, P., Baars, H., Borrmann, S., Chen, Q., Farmer, D. K., Guenther, A., Gunthe, S. S., Jimenez, J. L., Karl, T., Longo, K., Manzi, A., Müller, T., Pauliquevis, T., Petters, M. D., Prenni, A. J., Pöschl, U., Rizzo, L. V., Schneider, J., Smith, J. N., Swietlicki, E., Tota, J., Wang, J., Wiedensohler, A., and Zorn, S. R.: An overview of the Amazonian Aerosol Characterization Experiment 2008 (AMAZE-08), Atmos. Chem. Phys., 10, 11415–11438, https://doi.org/10.5194/acp-10-11415-2010, 2010a.
Martin, S. T., Andreae, M. O., Artaxo, P., Baumgardner, D., Chen, Q., Goldstein, A. H., Guenther, A., Heald, C. L., Mayol-Bracero, O. L., McMurry, P. H., Pauliquevis, T., Poschl, U., Prather, K. A., Roberts, G. C., Saleska, S. R., Dias, M. A. S., Spracklen, D. V., Swietlicki, E., and Trebs, I.: Sources and Properties of Amazonian Aerosol Particles, Rev. Geophys., 48, Rg2002, https://doi.org/10.1029/2008rg000280, 2010b.
Martin, S. T., Artaxo, P., Machado, L. A. T., Manzi, A. O., Souza, R. A. F., Schumacher, C., Wang, J., Andreae, M. O., Barbosa, H. M. J., Fan, J., Fisch, G., Goldstein, A. H., Guenther, A., Jimenez, J. L., Pöschl, U., Silva Dias, M. A., Smith, J. N., and Wendisch, M.: Introduction: Observations and Modeling of the Green Ocean Amazon (GoAmazon2014/5), Atmos. Chem. Phys., 16, 4785–4797, https://doi.org/10.5194/acp-16-4785-2016, 2016.
Martin, S. T., Artaxo, P., Machado, L., Manzi, A. O., Souza, R. A. F., Schumacher, C., Wang, J., Biscaro, T., Brito, J., Calheiros, A., Jardine, K., Medeiros, A., Portela, B., de Sa, S. S., Adachi, K., Aiken, A. C., Albrecht, R., Alexander, L., Andreae, M. O., Barbosa, M. J., Buseck, P., Chand, D., Comstock, J. M., Day, D. A., Dubey, M., Fan, J., Fast, J., Fisch, G., Fortner, E., Giangrande, S., Gilles, M., Goldststein, A. H., Guenther, A., Hubbe, J., Jensen, M., Jimenez, J. L., Keutsch, F. N., Kim, S., Kuang, C., Laskskin, A., McKinney, K., Mei, F., Miller, M., Nascimento, R., Pauliquevis, T., Pekour, M., Peres, J., Petaja, T., Pohlker, C., Poschl, U., Rizzo, L., Schmid, B., Shilling, J. E., Dias, M. A. S., Smith, J. N., Tomlmlinson, J. M., Tota, J., and Wendisch, M.: The Green Ocean Amazon Experiment (Goamazon2014/5) Observes Pollution Affecting Gases, Aerosols, Clouds, and Rainfall over the Rain Forest, B. Am. Meteorol. Soc., 98, 981–997, https://doi.org/10.1175/Bams-D-15-00221.1, 2017.
Medeiros, A. S. S., Calderaro, G., Guimaraes, P. C., Magalhaes, M. R., Morais, M. V. B., Rafee, S. A. A., Ribeiro, I. O., Andreoli, R. V., Martins, J. A., Martins, L. D., Martin, S. T., and Souza, R. A. F.: Power plant fuel switching and air quality in a tropical, forested environment, Atmos. Chem. Phys., 17, 8987–8998, https://doi.org/10.5194/acp-17-8987-2017, 2017.
Morgan, W. T., Allan, J. D., Flynn, M., Darbyshire, E., Hodgson, A., Johnson, T., Haywood, J. M., Freitas, S., Longo, K., Artaxo, P., and Coe, H.: Overview of the South American Biomass Burning Analysis (SAMBBA) Field Experiment, in: Nucleation and Atmospheric Aerosols, edited by: DeMott, P. J. and Odowd, C. D., AIP Conference Proceedings, 23–28 June 2013, Fort Collins, Colorado, USA, Amer. Inst. Physics, Melville, 587–590, 2013.
Ng, N. L., Canagaratna, M. R., Zhang, Q., Jimenez, J. L., Tian, J., Ulbrich, I. M., Kroll, J. H., Docherty, K. S., Chhabra, P. S., Bahreini, R., Murphy, S. M., Seinfeld, J. H., Hildebrandt, L., Donahue, N. M., DeCarlo, P. F., Lanz, V. A., Prévôt, A. S. H., Dinar, E., Rudich, Y., and Worsnop, D. R.: Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry, Atmos. Chem. Phys., 10, 4625–4641, https://doi.org/10.5194/acp-10-4625-2010, 2010.
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017.
Nguyen, T. B., Coggon, M. M., Bates, K. H., Zhang, X., Schwantes, R. H., Schilling, K. A., Loza, C. L., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Organic aerosol formation from the reactive uptake of isoprene epoxydiols (IEPOX) onto non-acidified inorganic seeds, Atmos. Chem. Phys., 14, 3497–3510, https://doi.org/10.5194/acp-14-3497-2014, 2014.
Odum, J. R., Hoffmann, T., Bowman, F., Collins, D., Flagan, R. C., and Seinfeld, J. H.: Gas/particle partitioning and secondary organic aerosol yields, Environ. Sci. Technol., 30, 2580–2585, https://doi.org/10.1021/es950943+, 1996.
Paatero, P.: Least squares formulation of robust non-negative factor analysis, Chemometrics and Intelligent Laboratory Systems, 37, 23–35, https://doi.org/10.1016/S0169-7439(96)00044-5, 1997.
Paatero, P. and Tapper, U.: Positive matrix factorization: A non-negative factor model with optimal utilization of error estimates of data values, Environmetrics, 5, 111–126, https://doi.org/10.1002/env.3170050203, 1994.
Parrish, D. D., Hahn, C. J., Williams, E. J., Norton, R. B., Fehsenfeld, F. C., Singh, H. B., Shetter, J. D., Gandrud, B. W., and Ridley, B. A.: Indications Of Photochemical Histories Of Pacific Air Masses From Measurements Of Atmospheric Trace Species At Point Arena, California, J. Geophys. Res.-Atmos., 97, 15883–15901, https://doi.org/10.1029/92jd01242, 1992.
Pierce, J. R., Leaitch, W. R., Liggio, J., Westervelt, D. M., Wainwright, C. D., Abbatt, J. P. D., Ahlm, L., Al-Basheer, W., Cziczo, D. J., Hayden, K. L., Lee, A. K. Y., Li, S.-M., Russell, L. M., Sjostedt, S. J., Strawbridge, K. B., Travis, M., Vlasenko, A., Wentzell, J. J. B., Wiebe, H. A., Wong, J. P. S., and Macdonald, A. M.: Nucleation and condensational growth to CCN sizes during a sustained pristine biogenic SOA event in a forested mountain valley, Atmos. Chem. Phys., 12, 3147–3163, https://doi.org/10.5194/acp-12-3147-2012, 2012.
Riipinen, I., Pierce, J. R., Yli-Juuti, T., Nieminen, T., Häkkinen, S., Ehn, M., Junninen, H., Lehtipalo, K., Petäjä, T., Slowik, J., Chang, R., Shantz, N. C., Abbatt, J., Leaitch, W. R., Kerminen, V.-M., Worsnop, D. R., Pandis, S. N., Donahue, N. M., and Kulmala, M.: Organic condensation: a vital link connecting aerosol formation to cloud condensation nuclei (CCN) concentrations, Atmos. Chem. Phys., 11, 3865–3878, https://doi.org/10.5194/acp-11-3865-2011, 2011.
Riipinen, I., Yli-Juuti, T., Pierce, J. R., Petaja, T., Worsnop, D. R., Kulmala, M., and Donahue, N. M.: The contribution of organics to atmospheric nanoparticle growth, Nat. Geosci., 5, 453–458, https://doi.org/10.1038/ngeo1499, 2012.
Robinson, A. L., Donahue, N. M., Shrivastava, M. K., Weitkamp, E. A., Sage, A. M., Grieshop, A. P., Lane, T. E., Pierce, J. R., and Pandis, S. N.: Rethinking organic aerosols: semivolatile emissions and photochemical aging, Science, 315, 1259–1262, https://doi.org/10.1126/science.1133061, 2007.
Robinson, N. H., Hamilton, J. F., Allan, J. D., Langford, B., Oram, D. E., Chen, Q., Docherty, K., Farmer, D. K., Jimenez, J. L., Ward, M. W., Hewitt, C. N., Barley, M. H., Jenkin, M. E., Rickard, A. R., Martin, S. T., McFiggans, G., and Coe, H.: Evidence for a significant proportion of Secondary Organic Aerosol from isoprene above a maritime tropical forest, Atmos. Chem. Phys., 11, 1039–1050, https://doi.org/10.5194/acp-11-1039-2011, 2011.
Sage, A. M., Weitkamp, E. A., Robinson, A. L., and Donahue, N. M.: Evolving mass spectra of the oxidized component of organic aerosol: results from aerosol mass spectrometer analyses of aged diesel emissions, Atmos. Chem. Phys., 8, 1139–1152, https://doi.org/10.5194/acp-8-1139-2008, 2008.
Schichtel, B. A., Malm, W. C., Bench, G., Fallon, S., McDade, C. E., Chow, J. C., and Watson, J. G.: Fossil and contemporary fine particulate carbon fractions at 12 rural and urban sites in the United States, J. Geophys. Res.-Atmos., 113, D02311, https://doi.org/10.1029/2007jd008605, 2008.
Schulz, C., Schneider, J., Amorim Holanda, B., Appel, O., Costa, A., de Sá, S. S., Dreiling, V., Fütterer, D., Jurkat-Witschas, T., Klimach, T., Krämer, M., Martin, S. T., Mertes, S., Pöhlker, M. L., Sauer, D., Voigt, C., Weinzierl, B., Ziereis, H., Zöger, M., Andreae, M. O., Artaxo, P., Machado, L. A. T., Pöschl, U., Wendisch, M., and Borrmann, S.: Aircraft-based observations of isoprene epoxydiol-derived secondary organic aerosol (IEPOX-SOA) in the tropical upper troposphere over the Amazon region, Atmos. Chem. Phys. Discuss., https://doi.org/10.5194/acp-2018-232, in review, 2018.
Scott, C. E., Spracklen, D. V., Pierce, J. R., Riipinen, I., D'Andrea, S. D., Rap, A., Carslaw, K. S., Forster, P. M., Artaxo, P., Kulmala, M., Rizzo, L. V., Swietlicki, E., Mann, G. W., and Pringle, K. J.: Impact of gas-to-particle partitioning approaches on the simulated radiative effects of biogenic secondary organic aerosol, Atmos. Chem. Phys., 15, 12989–13001, https://doi.org/10.5194/acp-15-12989-2015, 2015.
Setyan, A., Zhang, Q., Merkel, M., Knighton, W. B., Sun, Y., Song, C., Shilling, J. E., Onasch, T. B., Herndon, S. C., Worsnop, D. R., Fast, J. D., Zaveri, R. A., Berg, L. K., Wiedensohler, A., Flowers, B. A., Dubey, M. K., and Subramanian, R.: Characterization of submicron particles influenced by mixed biogenic and anthropogenic emissions using high-resolution aerosol mass spectrometry: results from CARES, Atmos. Chem. Phys., 12, 8131–8156, https://doi.org/10.5194/acp-12-8131-2012, 2012.
Shilling, J. E., Zaveri, R. A., Fast, J. D., Kleinman, L., Alexander, M. L., Canagaratna, M. R., Fortner, E., Hubbe, J. M., Jayne, J. T., Sedlacek, A., Setyan, A., Springston, S., Worsnop, D. R., and Zhang, Q.: Enhanced SOA formation from mixed anthropogenic and biogenic emissions during the CARES campaign, Atmos. Chem. Phys., 13, 2091–2113, https://doi.org/10.5194/acp-13-2091-2013, 2013.
Shrivastava, M., Cappa, C. D., Fan, J. W., Goldstein, A. H., Guenther, A. B., Jimenez, J. L., Kuang, C., Laskin, A., Martin, S. T., Ng, N. L., Petaja, T., Pierce, J. R., Rasch, P. J., Roldin, P., Seinfeld, J. H., Shilling, J., Smith, J. N., Thornton, J. A., Volkamer, R., Wang, J., Worsnop, D. R., Zaveri, R. A., Zelenyuk, A., and Zhang, Q.: Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 55, 509–559, https://doi.org/10.1002/2016rg000540, 2017.
Slowik, J. G., Stroud, C., Bottenheim, J. W., Brickell, P. C., Chang, R. Y.-W., Liggio, J., Makar, P. A., Martin, R. V., Moran, M. D., Shantz, N. C., Sjostedt, S. J., van Donkelaar, A., Vlasenko, A., Wiebe, H. A., Xia, A. G., Zhang, J., Leaitch, W. R., and Abbatt, J. P. D.: Characterization of a large biogenic secondary organic aerosol event from eastern Canadian forests, Atmos. Chem. Phys., 10, 2825–2845, https://doi.org/10.5194/acp-10-2825-2010, 2010.
Sullivan, A. P. and Weber, R. J.: Chemical characterization of the ambient organic aerosol soluble in water: 2. Isolation of acid, neutral, and basic fractions by modified size-exclusion chromatography, J. Geophys. Res.-Atmos., 111, D05315, https://doi.org/10.1029/2005jd006486, 2006.
Sullivan, A. P., Peltier, R. E., Brock, C. A., de Gouw, J. A., Holloway, J. S., Warneke, C., Wollny, A. G., and Weber, R. J.: Airborne measurements of carbonaceous aerosol soluble in water over northeastern United States: Method development and an investigation into water-soluble organic carbon sources, J. Geophys. Res.-Atmos., 111, D23s46, https://doi.org/10.1029/2006jd007072, 2006.
Surratt, J. D., Chan, A. W., Eddingsaas, N. C., Chan, M., Loza, C. L., Kwan, A. J., Hersey, S. P., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Reactive intermediates revealed in secondary organic aerosol formation from isoprene, P. Natl. Acad. Sci. USA, 107, 6640–6645, https://doi.org/10.1073/pnas.0911114107, 2010.
Takegawa, N., Miyakawa, T., Kondo, Y., Blake, D. R., Kanaya, Y., Koike, M., Fukuda, M., Komazaki, Y., Miyazaki, Y., Shimono, A., and Takeuchi, T.: Evolution of submicron organic aerosol in polluted air exported from Tokyo, Geophys. Res. Lett., 33, L15814, https://doi.org/10.1029/2006gl025815, 2006.
Ulbrich, I. M., Canagaratna, M. R., Zhang, Q., Worsnop, D. R., and Jimenez, J. L.: Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data, Atmos. Chem. Phys., 9, 2891–2918, https://doi.org/10.5194/acp-9-2891-2009, 2009.
Volkamer, R., Jimenez, J. L., San Martini, F., Dzepina, K., Zhang, Q., Salcedo, D., Molina, L. T., Worsnop, D. R., and Molina, M. J.: Secondary organic aerosol formation from anthropogenic air pollution: Rapid and higher than expected, Geophys. Res. Lett., 33, L17811, https://doi.org/10.1029/2006gl026899, 2006.
Weber, R. J., Sullivan, A. P., Peltier, R. E., Russell, A., Yan, B., Zheng, M., de Gouw, J., Warneke, C., Brock, C., Holloway, J. S., Atlas, E. L., and Edgerton, E.: A study of secondary organic aerosol formation in the anthropogenic-influenced southeastern United States, J. Geophys. Res.-Atmos., 112, D13302, https://doi.org/10.1029/2007jd008408, 2007.
Xu, L., Guo, H. Y., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S. H., Nenes, A., Weber, R. J., and Ng, N. L.: Correction to Supporting Information for Xu et al., Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, E4509, https://doi.org/10.1073/pnas.1512279112, 2015.
Yokelson, R. J., Karl, T., Artaxo, P., Blake, D. R., Christian, T. J., Griffith, D. W. T., Guenther, A., and Hao, W. M.: The Tropical Forest and Fire Emissions Experiment: overview and airborne fire emission factor measurements, Atmos. Chem. Phys., 7, 5175–5196, https://doi.org/10.5194/acp-7-5175-2007, 2007.
Yokelson, R. J., Crounse, J. D., DeCarlo, P. F., Karl, T., Urbanski, S., Atlas, E., Campos, T., Shinozuka, Y., Kapustin, V., Clarke, A. D., Weinheimer, A., Knapp, D. J., Montzka, D. D., Holloway, J., Weibring, P., Flocke, F., Zheng, W., Toohey, D., Wennberg, P. O., Wiedinmyer, C., Mauldin, L., Fried, A., Richter, D., Walega, J., Jimenez, J. L., Adachi, K., Buseck, P. R., Hall, S. R., and Shetter, R.: Emissions from biomass burning in the Yucatan, Atmos. Chem. Phys., 9, 5785–5812, https://doi.org/10.5194/acp-9-5785-2009, 2009.
Zaveri, R. A., Easter, R. C., Shilling, J. E., and Seinfeld, J. H.: Modeling kinetic partitioning of secondary organic aerosol and size distribution dynamics: representing effects of volatility, phase state, and particle-phase reaction, Atmos. Chem. Phys., 14, 5153–5181, https://doi.org/10.5194/acp-14-5153-2014, 2014.
Zhang, Q., Alfarra, M. R., Worsnop, D. R., Allan, J. D., Coe, H., Canagaratna, M. R., and Jimenez, J. L.: Deconvolution and quantification of hydrocarbon-like and oxygenated organic aerosols based on aerosol mass spectrometry, Environ. Sci. Technol., 39, 4938–4952, 2005a.
Zhang, Q., Worsnop, D. R., Canagaratna, M. R., and Jimenez, J. L.: Hydrocarbon-like and oxygenated organic aerosols in Pittsburgh: insights into sources and processes of organic aerosols, Atmos. Chem. Phys., 5, 3289–3311, https://doi.org/10.5194/acp-5-3289-2005, 2005b.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Allan, J. D., Coe, H., Ulbrich, I., Alfarra, M. R., Takami, A., Middlebrook, A. M., Sun, Y. L., Dzepina, K., Dunlea, E., Docherty, K., DeCarlo, P. F., Salcedo, D., Onasch, T., Jayne, J. T., Miyoshi, T., Shimono, A., Hatakeyama, S., Takegawa, N., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Williams, P., Bower, K., Bahreini, R., Cottrell, L., Griffin, R. J., Rautiainen, J., Sun, J. Y., Zhang, Y. M., and Worsnop, D. R.: Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes, Geophys. Res. Lett., 34, L13801, https://doi.org/10.1029/2007gl029979, 2007.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Ulbrich, I. M., Ng, N. L., Worsnop, D. R., and Sun, Y.: Understanding atmospheric organic aerosols via factor analysis of aerosol mass spectrometry: a review, Anal. Bioanal. Chem., 401, 3045–3067, https://doi.org/10.1007/s00216-011-5355-y, 2011.
Zhou, S., Collier, S., Xu, J. Z., Mei, F., Wang, J., Lee, Y. N., Sedlacek, A. J., Springston, S. R., Sun, Y. L., and Zhang, Q.: Influences of upwind emission sources and atmospheric processing on aerosol chemistry and properties at a rural location in the Northeastern US, J. Geophys. Res.-Atmos., 121, 6049–6065, https://doi.org/10.1002/2015jd024568, 2016.